Přibližný čas: 90 minut
studijní Cíle:
- Popsat proces RNA-seq knihovny příprava
- Popsat Illumina sekvenování metoda
Úvod k RNA-seq
RNA-seq je vzrušující, experimentální technika, která je využívána k prozkoumání a/nebo kvantifikaci genové exprese v rámci nebo mezi podmínky.,
jak víme, geny poskytují pokyny k výrobě proteinů, které vykonávají určitou funkci v buňce. I když všechny buňky obsahují stejné sekvence DNA, svalové buňky jsou odlišné od nervových buněk a dalších typů buněk z důvodu různých genů, které jsou zapnuté v těchto buňkách a různých Rna a proteinů produkovaných.
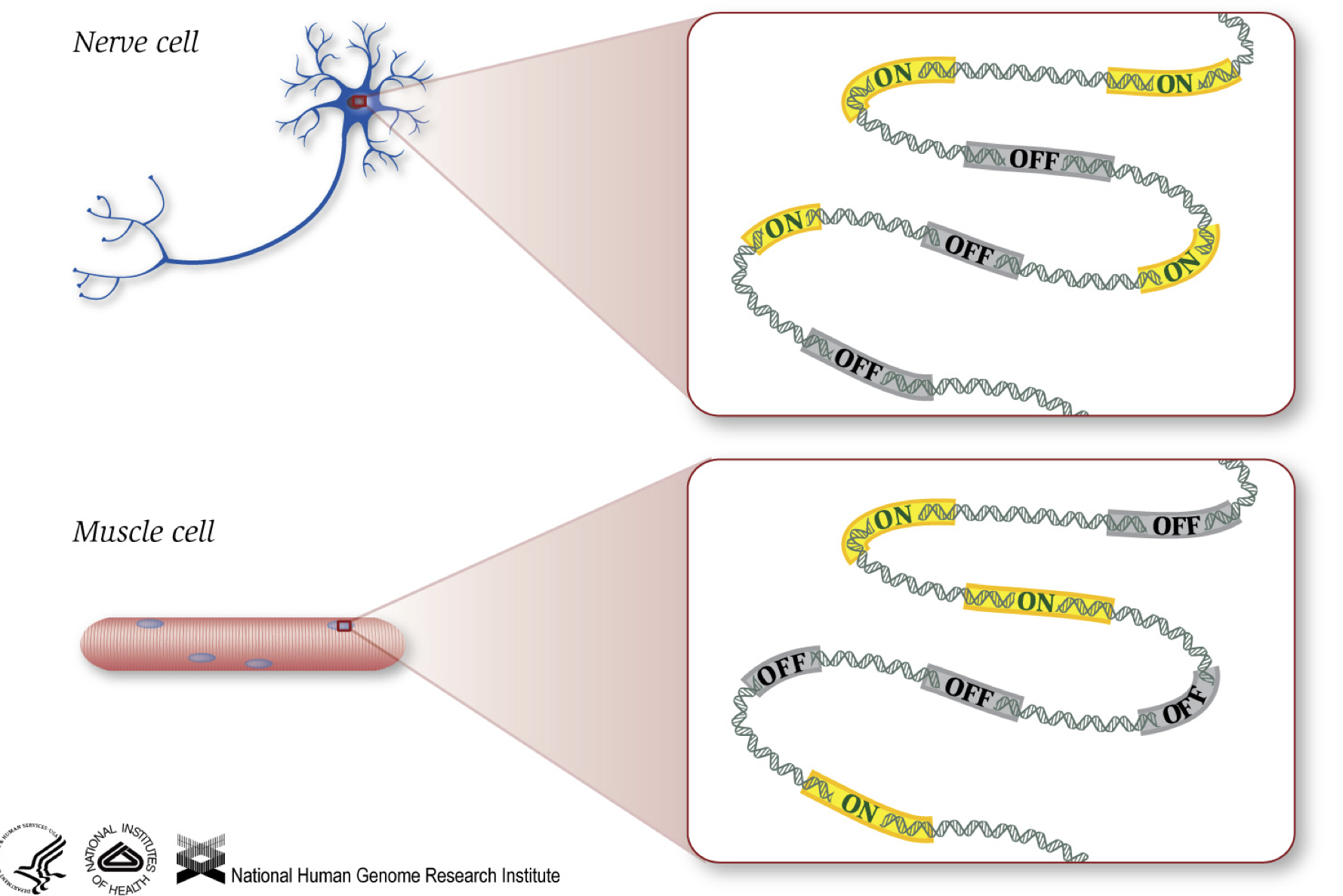
Různých biologických procesů, stejně jako mutace, může ovlivnit geny, které jsou zapnuté a které vypnuté, kromě toho, jak moc konkrétní geny jsou zapnuto/vypnuto.,
, Aby se proteiny, DNA je přepisována do mrna, nebo mRNA, která je přeložena do ribozomu do bílkovin. Některé geny však kódují RNA, která se nepřevádí do proteinu; tyto RNA se nazývají nekódující RNA nebo ncrna. Často tyto RNA mají funkci samy o sobě a zahrnují mimo jiné rRNAs, tRNAs a siRNAs. Všechny RNA přepisované z genů se nazývají přepisy.
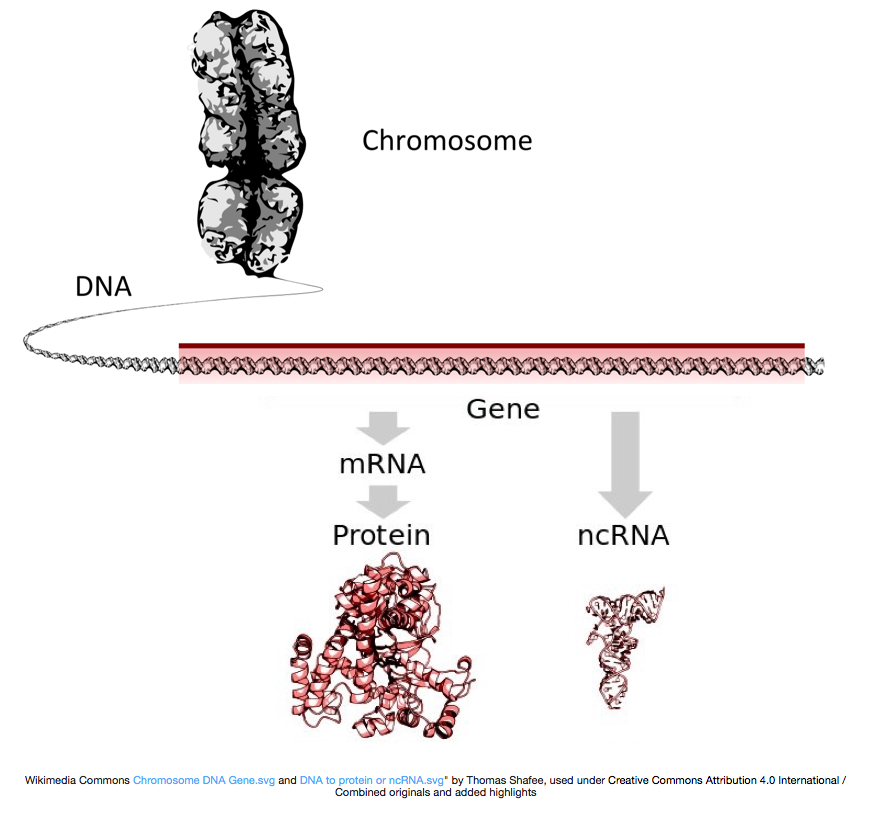
Aby bylo přeloženo do proteinů, RNA, musí projít zpracováním generovat mRNA., Na obrázku níže představuje horní pramen na obrázku gen v DNA, který se skládá z nepřeložených oblastí (UTRs) a otevřeného rámečku pro čtení. Geny jsou přepisovány do pre-mRNA, která stále obsahuje intronické sekvence. Po zpracování po transkripci se introny oddělí a přidá se ocas polyA a víčko 5′, aby se vytvořily zralé přepisy mRNA, které lze přeložit do proteinů.,
zatímco přepisy mRNA mají Polya ocas, mnoho nekódujících přepisů RNA není, protože post-transkripční zpracování se u těchto přepisů liší.
Transkriptomika
transkriptom je definován jako sbírka všech přepisových odečtů přítomných v buňce., RNA-seq dat mohou být použity k prozkoumání a/nebo vyčíslit transcriptome organismu, které mohou být využity pro následující typy experimentů:
- Diferenciální Genové Exprese: kvantitativní hodnocení a srovnání přepis úrovně
- Transcriptome montáž: budování profilu přepsal oblastí genomu, kvalitativní hodnocení.,
- Mohou být použity k pomoci vybudovat lepší genu modely a ověřit je pomocí sestavy
- Metatranscriptomics nebo společenství transcriptome analýzy
Illumina knihovna příprava
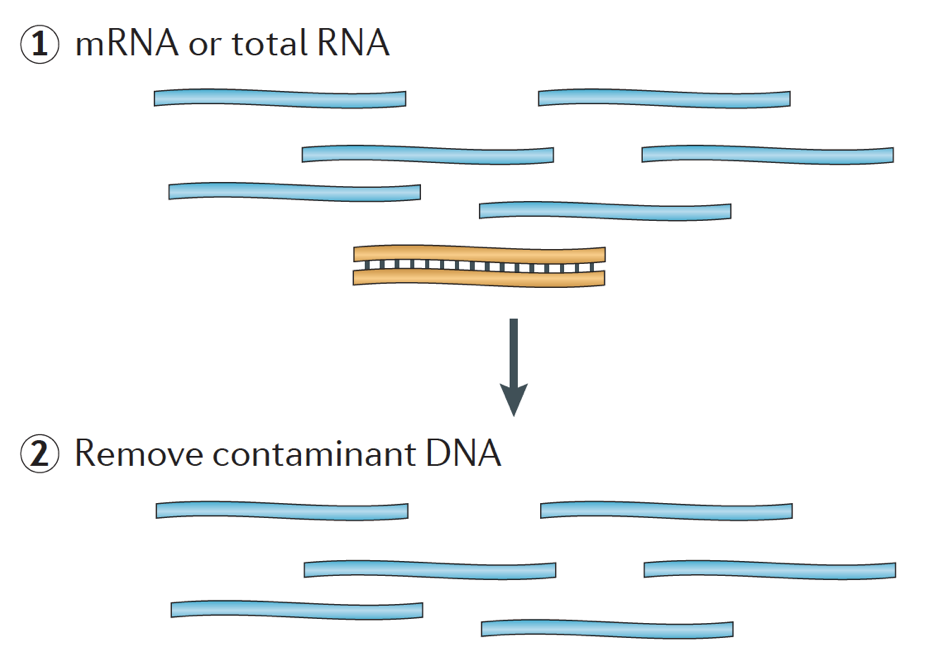
Při spuštění RNA-seq experiment, pro každý vzorek RNA musí být izolován a obrátil se do cDNA knihovny pro sekvenování. Obecný pracovní postup pro přípravu knihovny je podrobně popsán v podrobných obrázcích níže.
krátce se RNA izoluje ze vzorku a kontaminující DNA se odstraní Dnázou.,
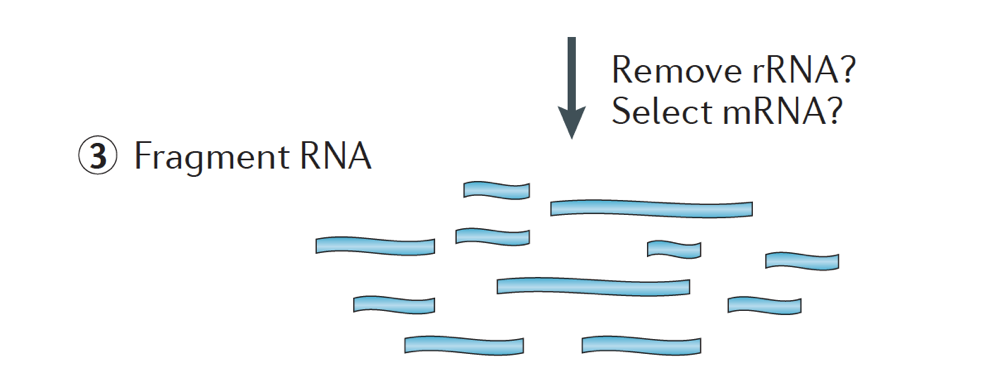
vzorek RNA pak podstoupí buď výběr mRNA (výběr polyA), nebo vyčerpání rRNA. Výsledná RNA je fragmentována.
Obecně platí, ribozomální RNA představuje většinu Rna přítomné v buňce, zatímco messenger RNAs představují malé procento z celkové RNA, ~2% u lidí. Pokud tedy chceme studovat geny kódující bílkoviny, musíme obohatit mRNA nebo vyčerpat rRNA., Pro analýzu diferenciální genové exprese je nejlepší obohatit o Poly(a)+, pokud se nesnažíte získat informace o dlouhých nekódujících RNA, pak proveďte depleci ribozomální RNA.
velikost cílových fragmentů v konečné knihovně je klíčovým parametrem pro konstrukci knihovny. Fragmentaci DNA se obvykle provádí fyzikální metody (tj. akustické stříhání a ultrazvuku) nebo enzymatické metody (tj., non-specifické endonukleázy koktejly a transposase tagmentation reakce.,
RNA je pak reverzní přepsal do dvouvláknové cDNA a sekvence adaptéry jsou pak přidány na konce fragmentů.
knihovny cDNA mohou být generovány tak, aby si uchovaly informace o tom, ze kterého řetězce DNA byla RNA přepsána. Knihovny, které si tyto informace uchovávají, se nazývají uvízlé knihovny, které jsou nyní standardem u sad Illumina TruSeq stranded RNA-Seq., Uvízlé knihovny by neměly být dražší než neřízené, takže ve skutečnosti neexistuje žádný důvod, proč tyto další informace nezískávat.,
k Dispozici jsou 3 typy cDNA knihoven k dispozici:
- Vpřed (secondstrand) – čte se podobají sekvence genu nebo secondstrand cDNA sekvence
- Reverse (firststrand) – čte se podobají doplňují genové sekvence nebo firststrand cDNA sekvence (TruSeq)
- Unstranded
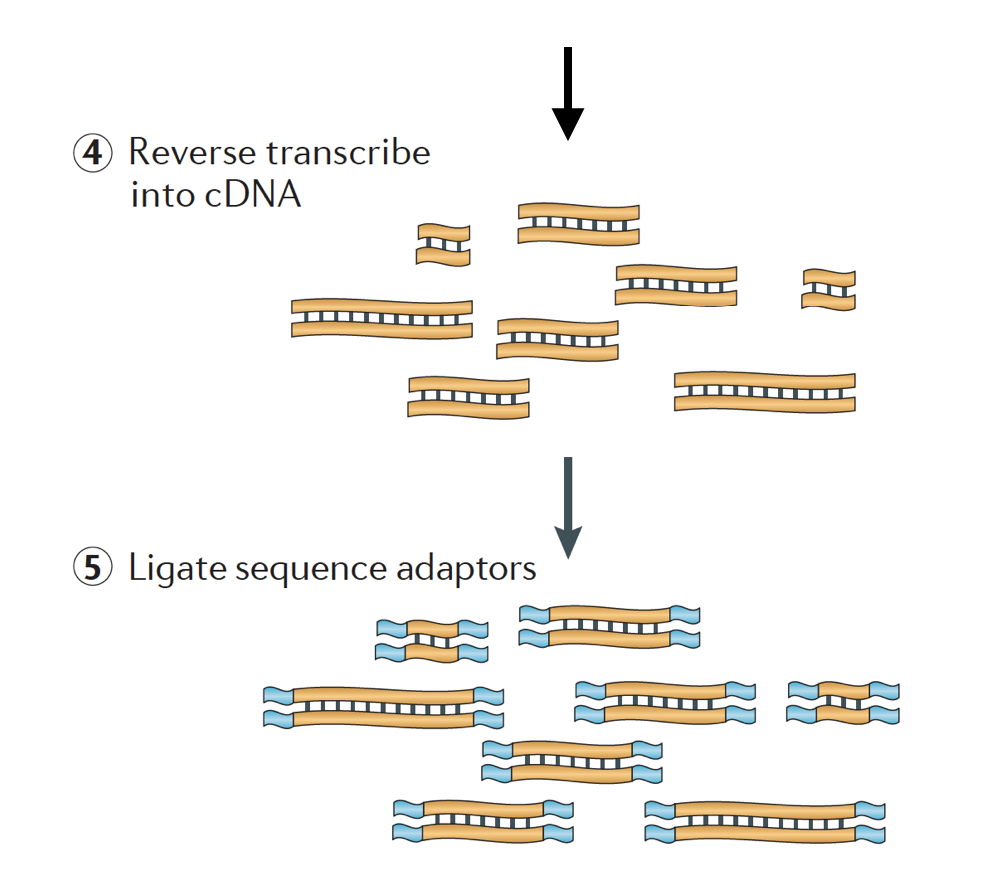
a Konečně, PCR fragmenty jsou zesíleny v případě potřeby, a fragmenty jsou vybrané velikosti (obvykle ~300-500bp), aby dokončit knihovna.
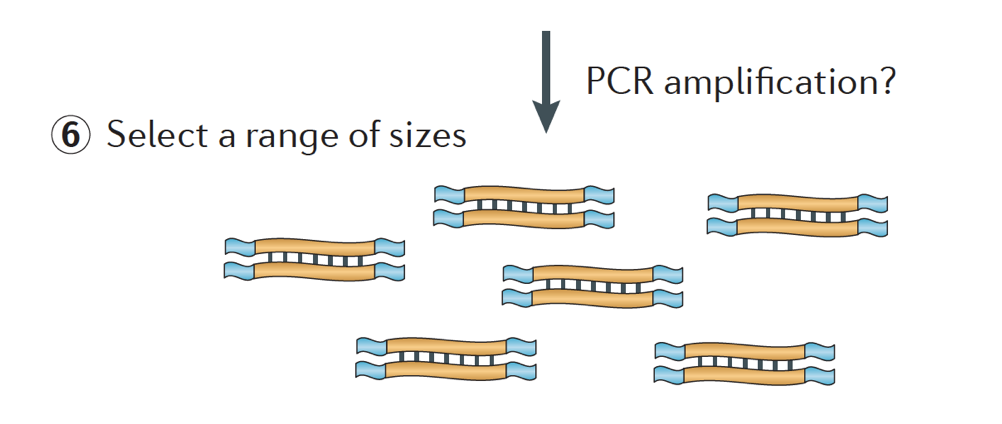
Image credit: Martin J. a. a Wang, Z., Nat. Rev., Genete. (2011) 12:671-682
Illumina Sekvenování
Single-end versus Spárované-end
Po přípravě knihovny, sekvenování může být provedena generovat nukleotidové sekvence na koncích fragmentů, které se nazývají čte. Budete mít na výběr sekvenování jeden konec fragmentů cDNA (single-end čte) nebo oba konce fragmentů (spárované-end čte).,
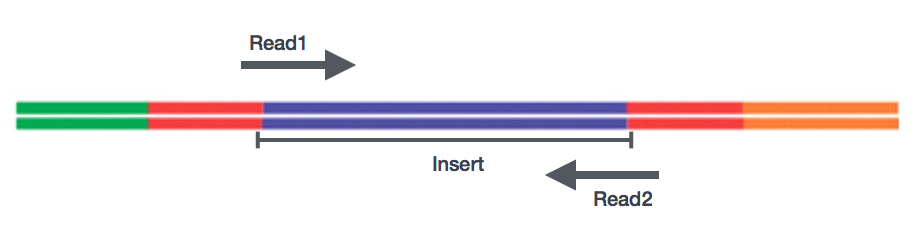
- SE – Jeden konec datového souboru => Pouze Read1
- PE – Párové-konec datového souboru => Read1 + Read2
- může být 2 samostatné FastQ soubory, nebo jen jeden s prokládané páry
Obecně single-end sekvenování je dostatečné, pokud se očekává, že čte bude odpovídat více místech v genomu (např. organismy s mnoha paralogní geny), sestavy jsou prováděny, nebo pro splice izoformy diferenciace. Uvědomte si, že párová koncová čtení jsou obecně 2x dražší.,
Různé platformy sekvenování
Existují různé platformy Illumina vybrat ze sekvence cDNA knihovny.

Image credit: Převzato z Illumina
Rozdíly v platformě se může změnit délka čte generované, kvalita čte, stejně jako celkový počet čte sekvence za běhu a množství čas potřebný k sekvenci knihovny., Různé platformy používají jinou průtokovou buňku, což je skleněný povrch potažený uspořádáním spárovaných oligos, které se doplňují s adaptéry přidanými do molekul šablony. Průtoková buňka je místem, kde dochází k sekvenačním reakcím.

Image credit: Převzato z Illumina
Sekvenování-do-syntéza
Illumina sekvenování technologie využívá sekvenování-do-syntéza přístup, který je popsán podrobněji níže.
v kroku jsou fragmenty DNA v knihovně cDNA denaturovány a aplikovány na skleněnou průtokovou buňku., Tyto denaturované fragmenty se vážou na komplementární oligos, které jsou již kovalentně vázány na pruhy průtokových buněk, což vede k připojení.
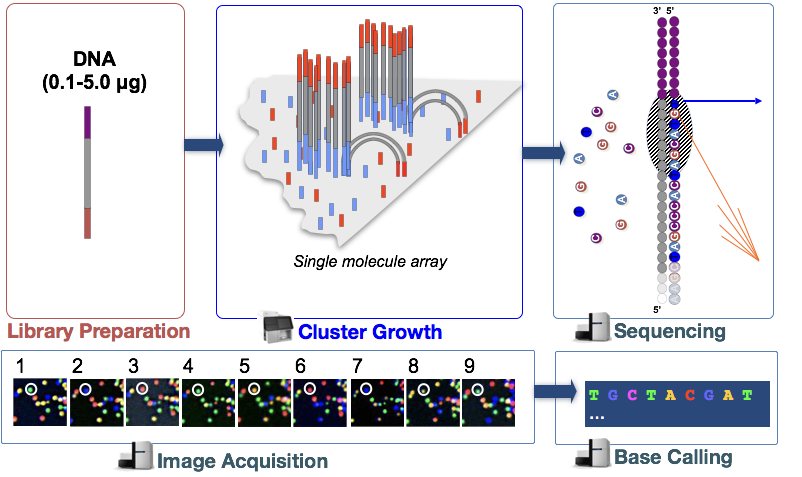
Clusteru Generace
Jakmile fragmenty připojený, fáze nazývá cluster generace začíná. Během tohoto kroku jsou jednotlivé fragmenty klonálně zesíleny, aby se vytvořil shluk (fragmenty v těsné blízkosti) identických fragmentů. To je nezbytné, aby fluorescence mohla být snadno zachycena z každé klastry namísto jediného fragmentu během inkorporace nukleotidů v dalším kroku.,
- Syntetizovat doplňují s polymeráza
- dsDNA je denaturována a původní DNA se promyje pryč odchodu syntetizované vlákno kovalentně vázán na tok buněk.
- jednovláknová hybridizace s přilehlým adaptérem pro vytvoření „můstku“
- dsDNA je rozšířena polymerázou. Každý pramen kovalentně vázán na jiný adaptér.
- opakujte mnohokrát, abyste klonálně zesílili všechny jedinečné fragmenty na průtokové buňce a vytvořili shluky identické sekvence.,
Sekvenování syntézou (& image acquisition)
Po clusteru generace, fluorescenčně značené nukleotidy jsou součástí jednoho najednou (cyklicky) a fluorescenční snímky jsou zachyceny určit, které nukleotidů dostane začleněny do každého clusteru v každém cyklu.
- denaturní klastry a konce bloku 3′, aby se zabránilo nežádoucímu primingu.
- hybridizovat sekvenční primery na adaptační sekvenci na volných koncích.,
- cyklus čtyři Ntps s fluorescenčními markery a terminátorovou sekvencí a polymerázami.
- jakmile je NTP začleněna, je cluster excitován světelným zdrojem a je vysílán charakteristický fluroscentní signál.
- barva je zaznamenána, pak se terminátor na barvivu rozštěpí a promyje. Proces se opakuje pro zadaný počet cyklů.,
Základní Volání
Illumina má proprietární software, který prochází všechny snímky pořízené v předchozí fázi a generuje textové soubory s sekvence informace o každém clusteru na základě fluorescence. Kromě volání základny, tento software přiřadí probablity skóre ukazují, jak jisté je to, o volání něco „A“, „T“, „G“ nebo „C“.
pokud existují nějaké nejasnosti, např., v určitém cyklu obraz pro cluster nemá výraznou barvu, která může být spojena s konkrétní nukleotidů, základní volání software bude mít nízkou pravděpodobnost, s ním spojené a přiřadit „N“ místo „A“, „T“, „G“ nebo „C“.
Na závěr,
- Počet shluků ~= Počet čtení
- Počet sekvenčních cyklů = Délka čte
počet cyklů (délka čte) bude záviset na sekvenční platformu používá, stejně jako vaše preference.
poznámka., Pokud chcete prozkoumat sekvenování syntézou do větší hloubky, doporučujeme tuto opravdu pěknou animaci dostupnou na Kanálu Youtube Illumina.
Multiplexování
v Závislosti na platformy Illumina (MiSeq, HiSeq, NextSeq), počet jízdních pruhů na toku buněk, a počet čtení, které lze získat v jednom jízdním pruhu, se velmi liší. Budete se muset rozhodnout, kolik čtení byste chtěli na vzorek (tj. hloubka sekvencování) a poté na základě platformy zvolíte vypočítat, kolik celkových pruhů budete potřebovat pro vaši sadu vzorků., Budeme hovořit více o úvahách při rozhodování v další lekci o experimentálních úvahách
typicky jsou poplatky za sekvenování za pruh průtokové buňky a budete moci spustit více vzorků za pruh. Illumina proto vymyslela pěknou multiplexní metodu, která umožňuje sdružovat a sekvenovat knihovny z několika vzorků současně ve stejném pruhu průtokové buňky. Tato metoda vyžaduje přidání indexů (v rámci adaptéru Illumina) nebo speciálních čárových kódů (mimo adaptér Illumina), jak je popsáno níže.,
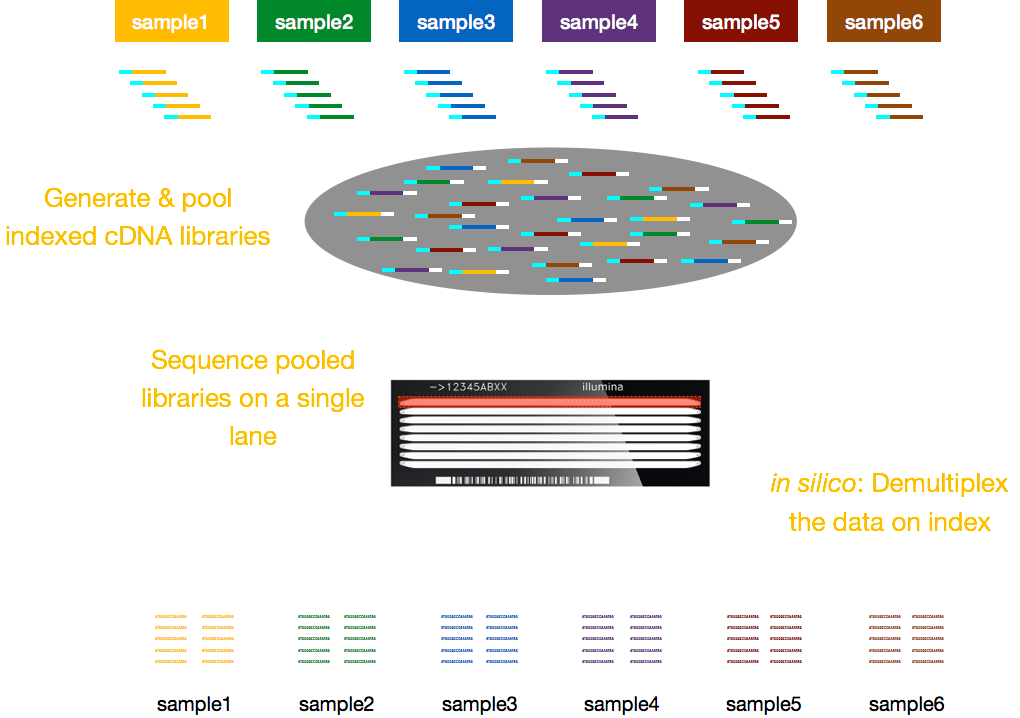
POZNÁMKA: pracovní postupy uvedené v této lekci je specifické pro Illumina sekvenování, které je v současné době nejvíce využívána metoda sekvenování., Ale tam jsou jiné dlouho-číst sekvenování metod stojí za zmínku, například:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomika: https://www.10xgenomics.com/
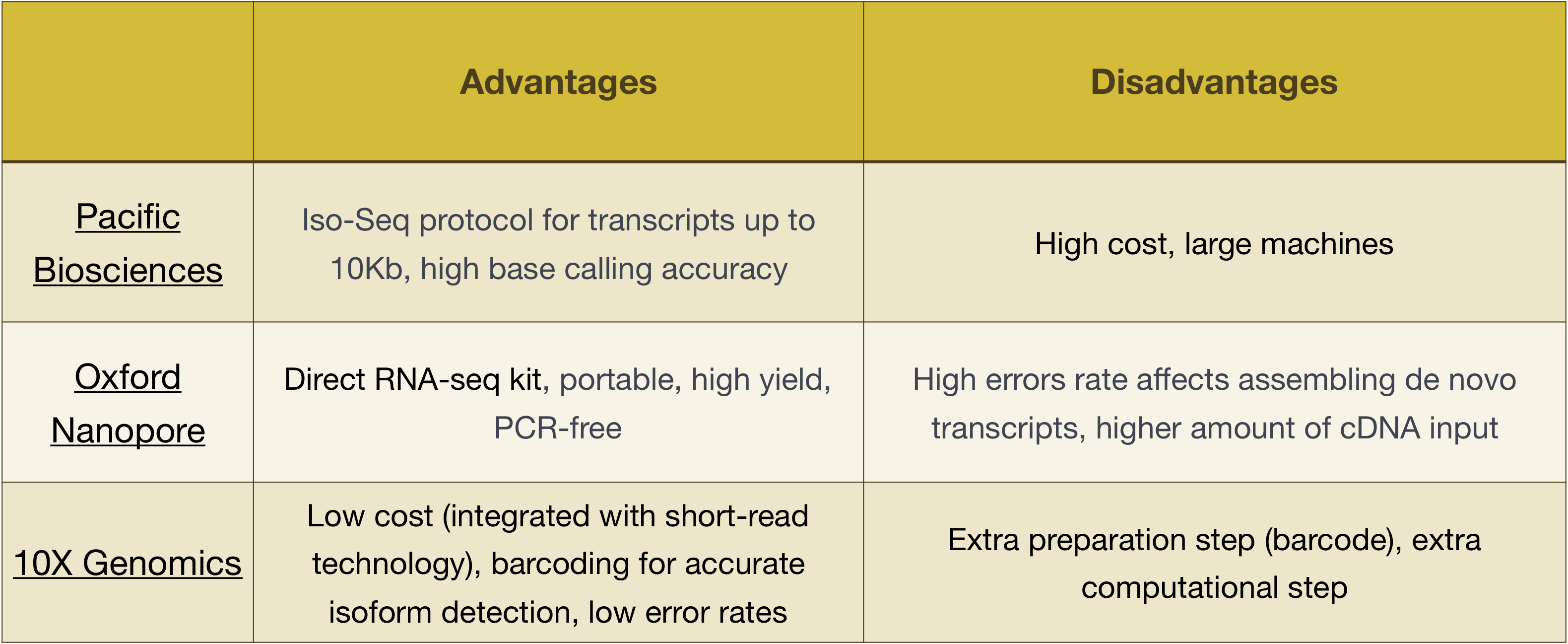
Výhody a nevýhody těchto technologií mohou být prozkoumány v následující tabulce:
Napsat komentář