Tiempo Aproximado: 90 minutos
objetivos de aprendizaje:
- describir el proceso de preparación de la biblioteca RNA-seq
- describir el método de secuenciación Illumina
Introducción a RNA-seq
emocionante técnica experimental que se utiliza para explorar y/o cuantificar la expresión génica dentro o entre Condiciones.,
como sabemos, los genes proporcionan instrucciones para producir proteínas, que realizan alguna función dentro de la célula. Aunque todas las células contienen la misma secuencia de ADN, las células musculares son diferentes de las células nerviosas y otros tipos de células debido a los diferentes genes que se activan en estas células y los diferentes ARN y proteínas producidas.
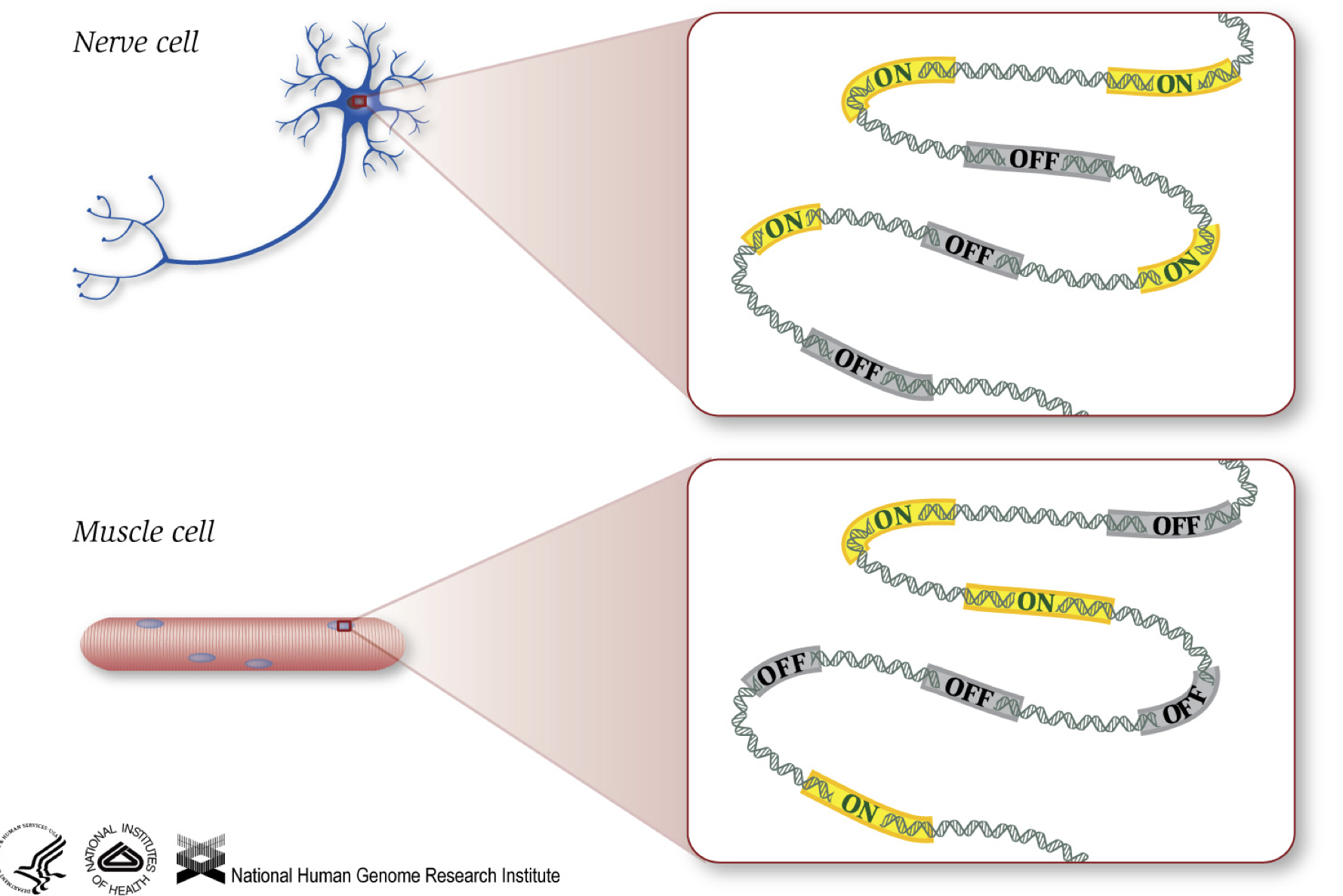
diferentes procesos biológicos, así como mutaciones, pueden afectar qué genes se activan y cuáles se desactivan, además de la cantidad de genes específicos que se activan / desactivan.,
para hacer proteínas, el ADN se transcribe en ARN mensajero, o ARNm, que es traducido por el ribosoma en proteína. Sin embargo, algunos genes codifican ARN que no se traduce en proteínas; estos ARN se denominan ARN no codificantes, o ncRNAs. A menudo estos ARN tienen una función en sí mismos e incluyen rRNAs, tRNAs y siRNAs, entre otros. Todos los ARN transcritos de genes se llaman transcripciones.
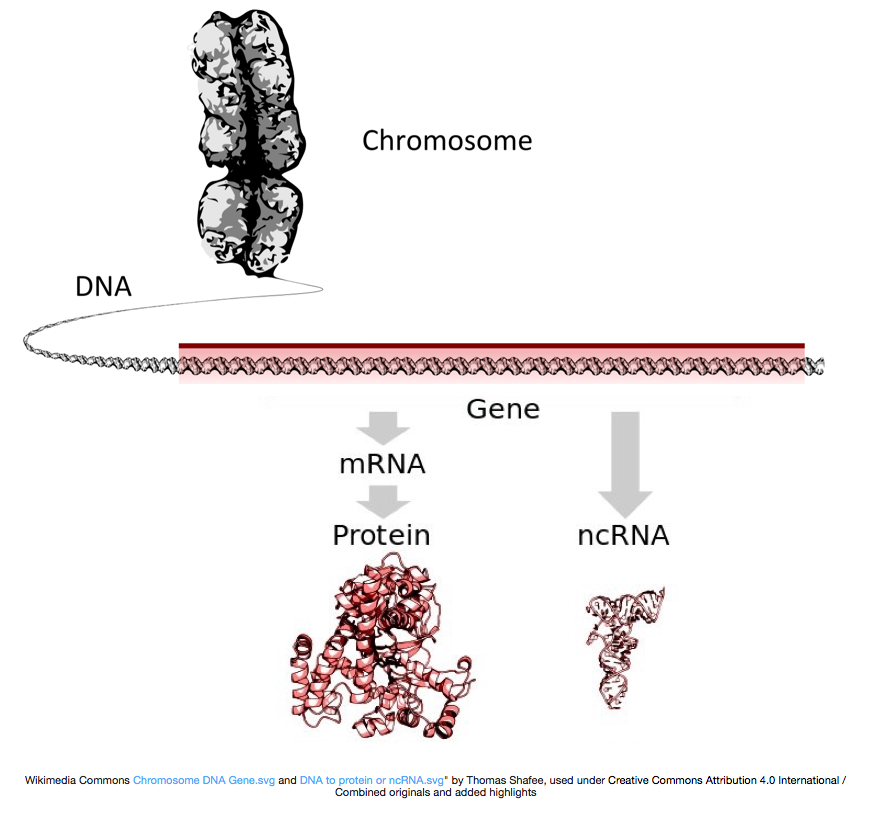
Para ser traducido a proteínas, el ARN debe sufrir una transformación para generar el arnm., En la siguiente figura, la hebra superior de la imagen representa un gen en el ADN, compuesto por las regiones no traducidas (UTRs) y el marco de lectura abierto. Los Genes se transcriben en pre-ARNm, que todavía contiene las secuencias intrónicas. Después del procesamiento post-transcipcional, los intrones se empalman y se agrega una cola de polyA y una tapa de 5′ para producir transcripciones maduras de ARNm, que se pueden traducir en proteínas.,
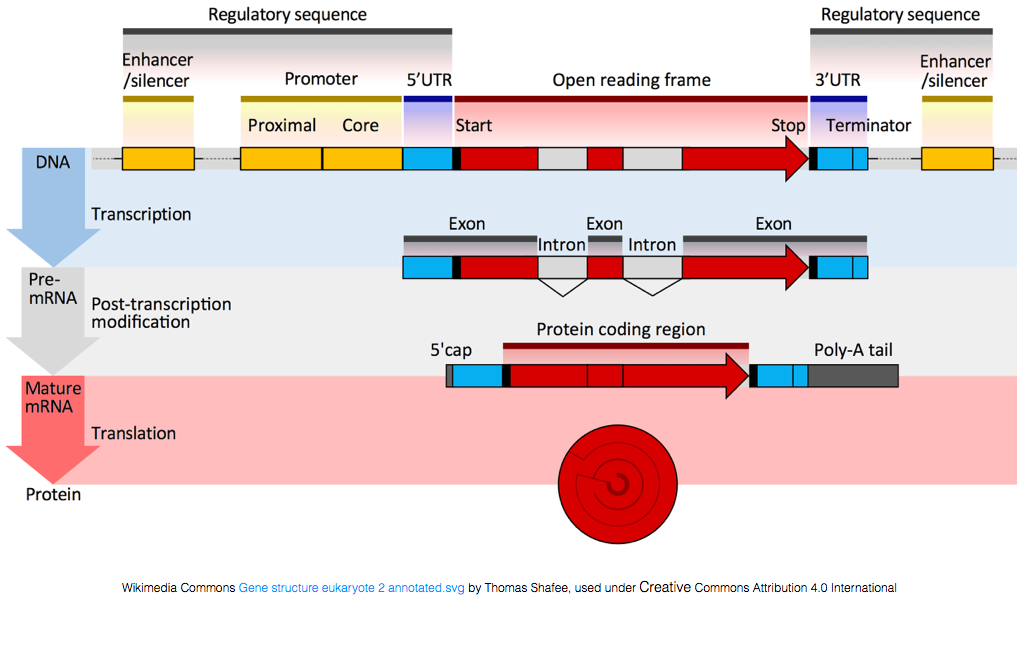
mientras que las transcripciones de ARNm tienen una cola de polyA, muchas de las transcripciones de ARN no codificantes no lo hacen ya que el procesamiento post-transcripcional es diferente para estas transcripciones.
transcriptómica
el transcriptoma se define como una colección de todas las lecturas de transcripción presentes en una célula., Los datos de RNA-seq pueden ser utilizados para explorar y/o cuantificar el transcriptoma de un organismo, el cual puede ser utilizado para los siguientes tipos de experimentos:
- expresión génica diferencial: evaluación cuantitativa y comparación de niveles de transcripción
- ensamblaje del transcriptoma: construyendo el perfil de regiones transcritas del genoma, una evaluación cualitativa.,
- Se puede usar para ayudar a construir mejores modelos de genes y verificarlos utilizando el ensamblado
- Metatranscriptómica o Análisis de transcriptoma comunitario
Illumina library preparation
al iniciar un experimento de RNA-seq, para cada muestra el RNA debe aislarse y convertirse en una biblioteca de ADNc para secuenciar. El flujo de trabajo general para la preparación de la biblioteca se detalla en las imágenes paso a paso a continuación.
brevemente, el ARN se aísla de la muestra y el ADN contaminante se elimina con DNasa.,
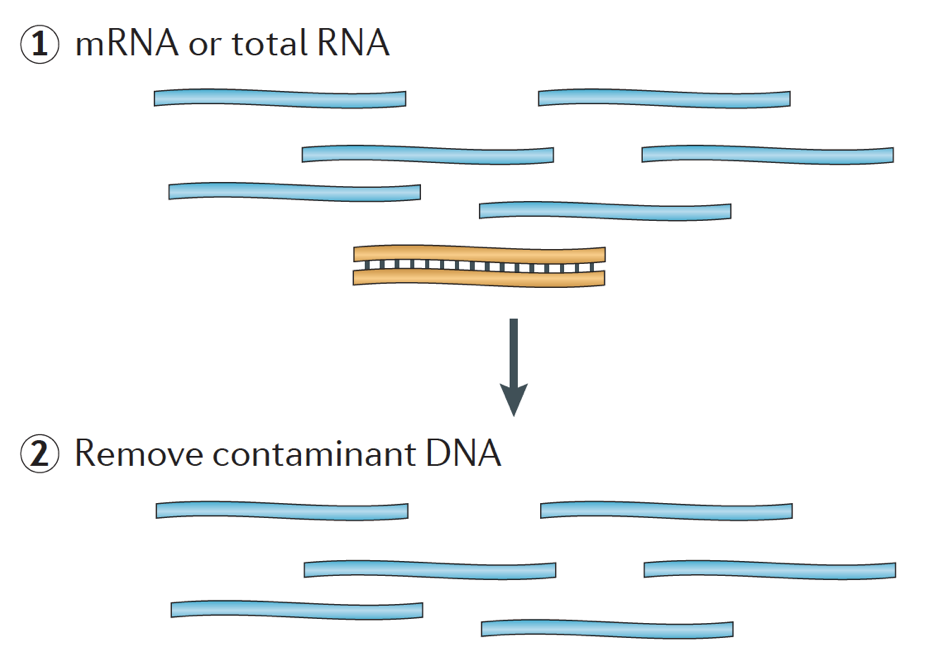
la muestra de ARN se somete a la selección del ARNm (selección polyA) o el agotamiento del ARNr. El ARN resultante está fragmentado.
generalmente, el ARN ribosomal representa la mayoría de los ARN presentes en una célula, mientras que los ARN mensajeros representan un pequeño porcentaje del ARN total, ~2% en humanos. Por lo tanto, si queremos estudiar los genes codificadores de proteínas, necesitamos enriquecer para ARNm o agotar el ARNr., Para el análisis diferencial de expresión génica, lo mejor es enriquecer para poli(a)+, a menos que esté apuntando a obtener información sobre ARN largos no codificantes, y luego hacer una depleción de ARN ribosomal.
El tamaño de los fragmentos de destino en la biblioteca final es un parámetro clave para la construcción de la biblioteca. La fragmentación del ADN se realiza típicamente por métodos físicos (es decir, cizallamiento acústico y sonicación) o métodos enzimáticos (es decir, cócteles de endonucleasas inespecíficas y reacciones de etiquetado de transposasa.,
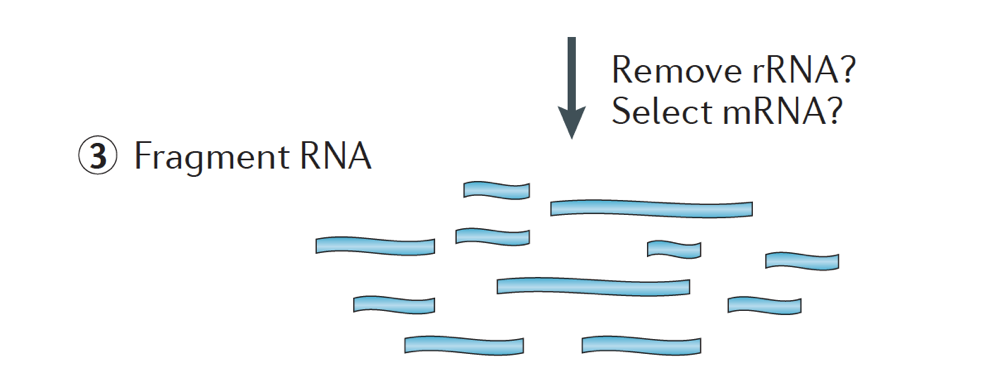
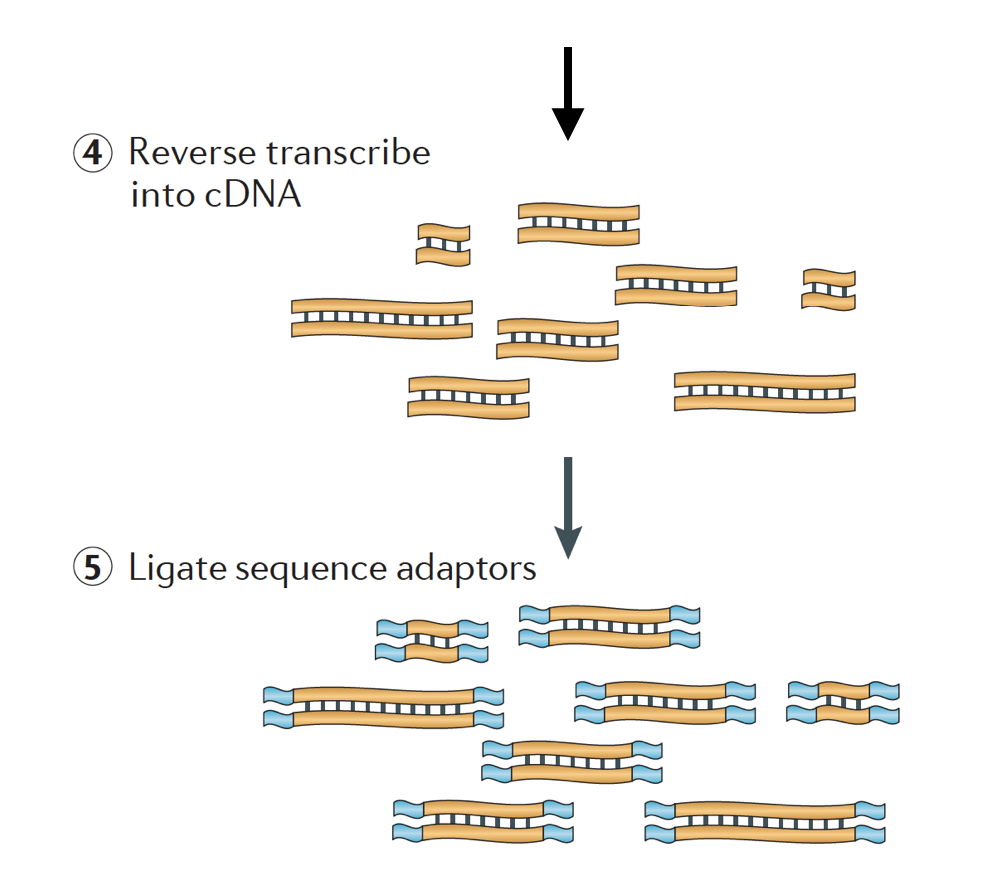
el ARN se transcribe a la inversa en cDNA de doble cadena y los adaptadores de secuencia se agregan a los extremos de los fragmentos.
Las bibliotecas de ADNc se pueden generar de manera que retengan información sobre de qué cadena de ADN se transcribió el ARN. Las bibliotecas que retienen esta información se llaman bibliotecas stranded, que ahora son estándar con los kits TruSeq stranded RNA-Seq de Illumina., Las bibliotecas varadas no deben ser más caras que las no etiquetadas, por lo que no hay realmente ninguna razón para no adquirir esta información adicional.,
Hay 3 tipos de bibliotecas de ADNc disponibles:
- Forward (secondstrand) – reads like the gene sequence or the secondstrand cDNA sequence
- Reverse (firststrand) – reads like the complement of the gene sequence or firststrand cDNA sequence (TruSeq)
- Unstranded
finalmente, los fragmentos se amplifican por PCR si es necesario, y los fragmentos se seleccionan de tamaño (generalmente ~300-500bp) para terminar la biblioteca.
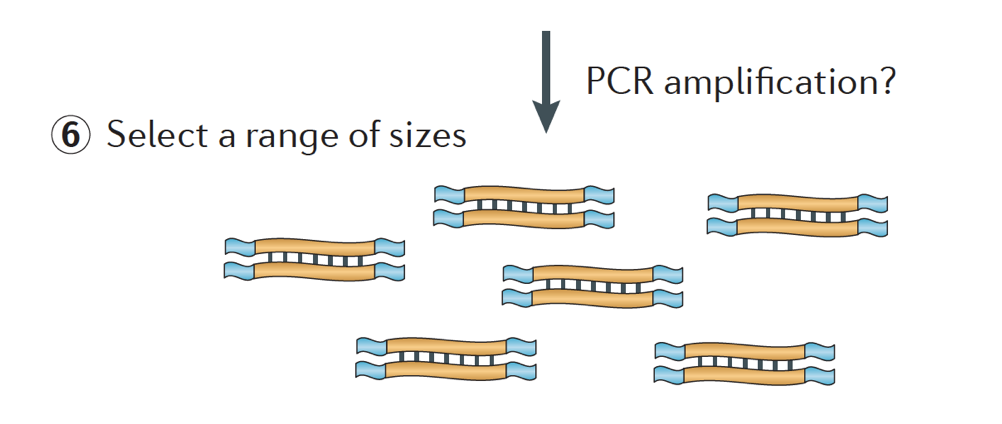
crédito de la imagen: Martin J. A. and Wang Z., Nat. Apo., Genet. (2011) 12:671-682
Illumina Sequencing
Single-end versus Paired-end
después de la preparación de las bibliotecas, se puede realizar la secuenciación para generar las secuencias de nucleótidos de los extremos de los fragmentos, que se denominan lecturas. Tendrá la opción de secuenciar un solo extremo de los fragmentos de ADNc (lecturas de un solo extremo) o ambos extremos de los fragmentos (lecturas de extremos emparejados).,
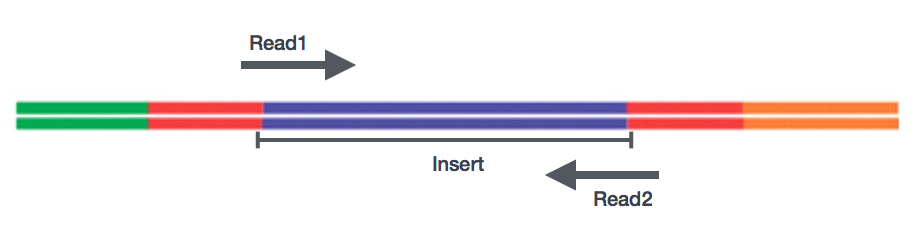
- SE – Single end dataset => solo Read1
- PE – pared-end dataset => Read1 + Read2
- archivos FASTQ o solo uno con pares intercalados
generalmente la secuenciación de un solo extremo es suficiente a menos que se espere que las lecturas coincidan con múltiples ubicaciones en el genoma (por ejemplo, organismos con muchos genes paralógicos), se están realizando ensamblajes o para la diferenciación de isoformas de empalme. Tenga en cuenta que las lecturas de extremo emparejado generalmente son 2 veces más caras.,
diferentes plataformas de secuenciación
hay una variedad de plataformas Illumina para elegir para secuenciar las bibliotecas de cDNA.

crédito de la imagen: adaptado de Illumina
Las diferencias en la plataforma pueden alterar la longitud de las lecturas generadas, la calidad de las lecturas, así como el número total de lecturas secuenciadas por ejecución y la cantidad de tiempo requerido para secuenciar las bibliotecas., Cada una de las diferentes plataformas utiliza una celda de flujo diferente, que es una superficie de vidrio recubierta con una disposición de oligos emparejados que son complementarios a los adaptadores agregados a sus moléculas de plantilla. La celda de flujo es donde tienen lugar las reacciones de secuenciación.

crédito de la imagen: adaptado de Illumina
secuenciación por síntesis
La tecnología de secuenciación Illumina utiliza un enfoque de secuenciación por síntesis que se describe con más detalle a continuación.
en el paso, los fragmentos de ADN en la biblioteca de ADNc se desnaturalizan y se aplican a la célula de flujo de vidrio., Estos fragmentos desnaturalizados se unen a los oligos complementarios que ya están covalentemente Unidos a los carriles de las celdas de flujo, lo que resulta en la Unión.
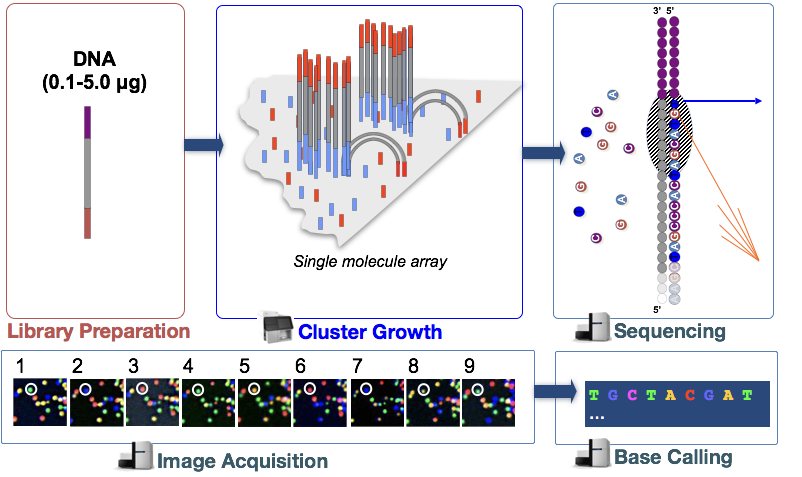
generación de clústeres
Una vez que los fragmentos se han unido, comienza una fase llamada Generación de clústeres. Durante este paso, los fragmentos individuales se amplifican clonalmente para crear un grupo (fragmentos en proximidad cercana) de fragmentos idénticos. Esto es necesario para que la fluorescencia pueda capturarse fácilmente de cada grupo, en lugar de un solo fragmento, durante la incorporación de nucleótidos en el siguiente paso.,
- sintetizar el complemento con polimerasa
- El dsDNA se desnaturaliza, y el ADN original se lava dejando la hebra sintetizada covalentemente unida a la célula de flujo.
- hebra simple se hibrida con el adaptador adyacente para formar un ‘puente’
- El dsDNA se extiende por la polimerasa. Cada hebra covalentemente unida a un adaptador diferente.
- repita muchas veces para amplificar clonalmente todos los fragmentos únicos en la celda de flujo para formar grupos de secuencia idéntica.,
secuenciación por síntesis (& adquisición de imágenes)
después de la generación del clúster, los nucleótidos marcados fluorescentemente se incorporan uno a la vez (cíclicamente) y se capturan imágenes de fluorescencia para identificar qué nucleótido se incorpora a cada clúster en cada ciclo.
- desnaturalizar los clústeres y el bloque 3′ termina para evitar el cebado no deseado.
- hibridar los cebadores de secuenciación a la secuencia del adaptador en los cabos sueltos.,
- Ciclo cuatro NTPs con marcadores fluorescentes y secuencia de terminadores y polimerasas.
- Una vez incorporado NTP, el clúster es excitado por una fuente de luz y se emite una señal característica de fluroscent.
- El color se registra, luego el terminador en el tinte se corta y se lava. El proceso se repite para un número específico de ciclos.,
Base Calling
Illumina tiene un software propietario que recorre todas las imágenes capturadas en la etapa anterior y genera archivos de texto con información de secuencia sobre cada clúster en función de la fluorescencia. Además de llamar a las bases, este software asigna una puntuación de probablidad para indicar qué tan seguro estaba sobre la llamada a algo una «A», una «T», una «G»o una «C».
Si hay ambigüedades, por ejemplo,, en un cierto ciclo, la imagen de un clúster no tiene un color distinto que pueda asociarse con un nucleótido específico, el software de llamada base tendrá una baja probabilidad asociada con él y asignará una «N» en lugar de «A», «T», «G» O «C».
En el cierre,
- Número de clústeres ~= Número de lecturas
- Número de ciclos de secuenciación = Longitud de lee
El número de ciclos (longitud de las lecturas) dependerá de la plataforma de secuenciación se utiliza, así como sus preferencias.
nota., Si desea explorar la secuenciación por síntesis en más profundidad, le recomendamos esta animación realmente agradable disponible en el canal de YouTube de Illumina.
multiplexación
dependiendo de la plataforma Illumina (MiSeq, HiSeq, NextSeq), el número de carriles por celda de flujo y el número de lecturas que se pueden obtener por carril varía ampliamente. Tendrá que decidir cuántas lecturas le gustaría por muestra (es decir, la profundidad de secuenciación) y luego, en función de la plataforma que elija, calcular cuántos carriles totales necesitará para su conjunto de muestras., Hablaremos más sobre las consideraciones al tomar esta decisión en la siguiente lección sobre consideraciones experimentales
normalmente, los cargos por secuenciación son por carril de la celda de flujo y podrá ejecutar múltiples muestras por carril. Illumina, por lo tanto, ha ideado un buen método de multiplexación que permite que las bibliotecas de varias muestras se agrupen y secuencien simultáneamente en el mismo carril de una celda de flujo. Este método requiere la adición de índices (dentro del adaptador Illumina) o códigos de barras especiales (fuera del adaptador Illumina) como se describe en el esquema a continuación.,
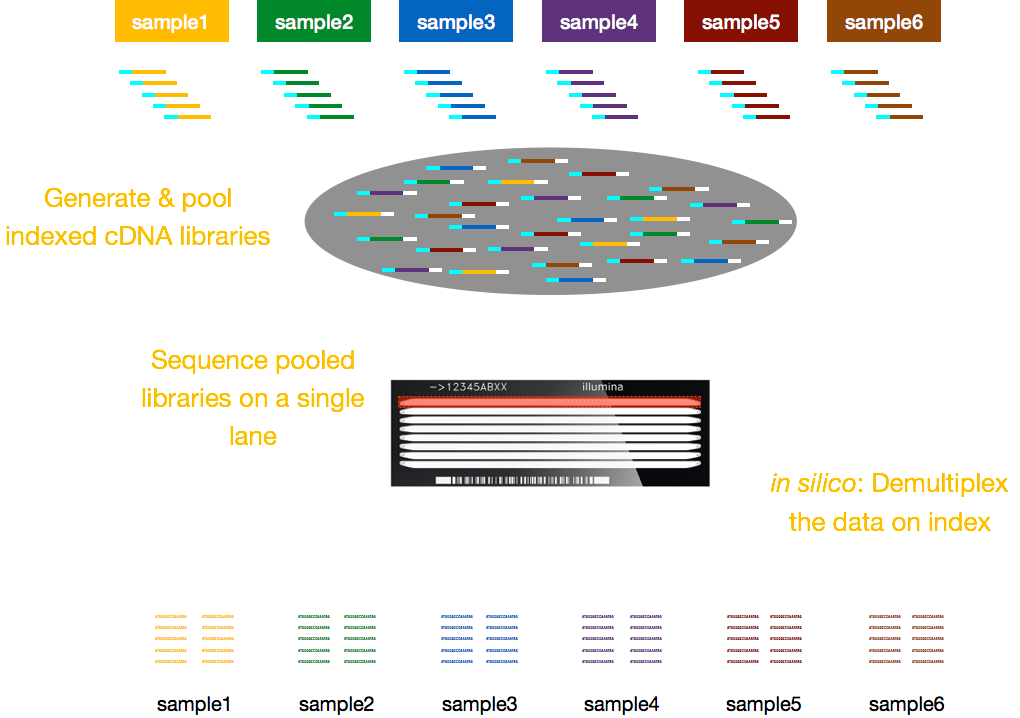
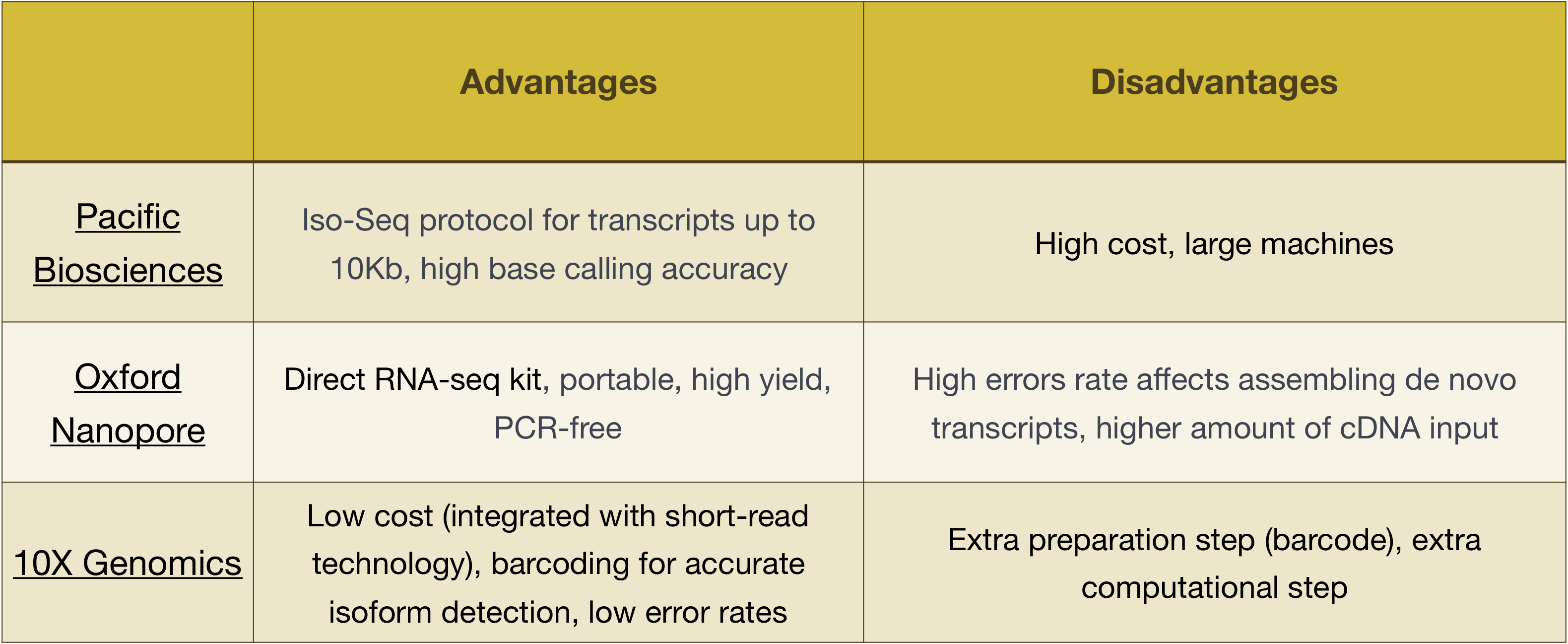
nota: el flujo de trabajo presentado en esta lección es específico para la secuenciación de Illumina, que es actualmente el método de secuenciación más utilizado., Pero hay otros métodos de secuenciación de larga lectura que vale la pena señalar, como:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10x Genomics: https://www.10xgenomics.com/
las ventajas y desventajas de estas tecnologías se pueden explorar en la siguiente tabla:
Deja una respuesta