Arvioitu aika: 90 minuuttia
Oppimisen Tavoitteet:
- Kuvata prosessi RNA-seq kirjasto valmistelu
- Kuvaile Illumina sekvensointi menetelmä
Johdanto-RNA-seq
RNA-seq on jännittävä kokeellinen tekniikka, jota käytetään tutkia ja/tai arvioida geenien ilmentyminen sisällä tai välillä ehtoja.,
kuten tiedämme, geenit antavat ohjeita proteiinien valmistamiseksi, jotka toimivat jonkin verran solun sisällä. Vaikka kaikki solut sisältävät saman DNA-sekvenssi -, lihas-solut ovat eri hermosolujen ja muita soluja, koska eri geenejä, jotka ovat käytössä näissä soluissa ja eri RNAs ja proteiineja tuotetaan.
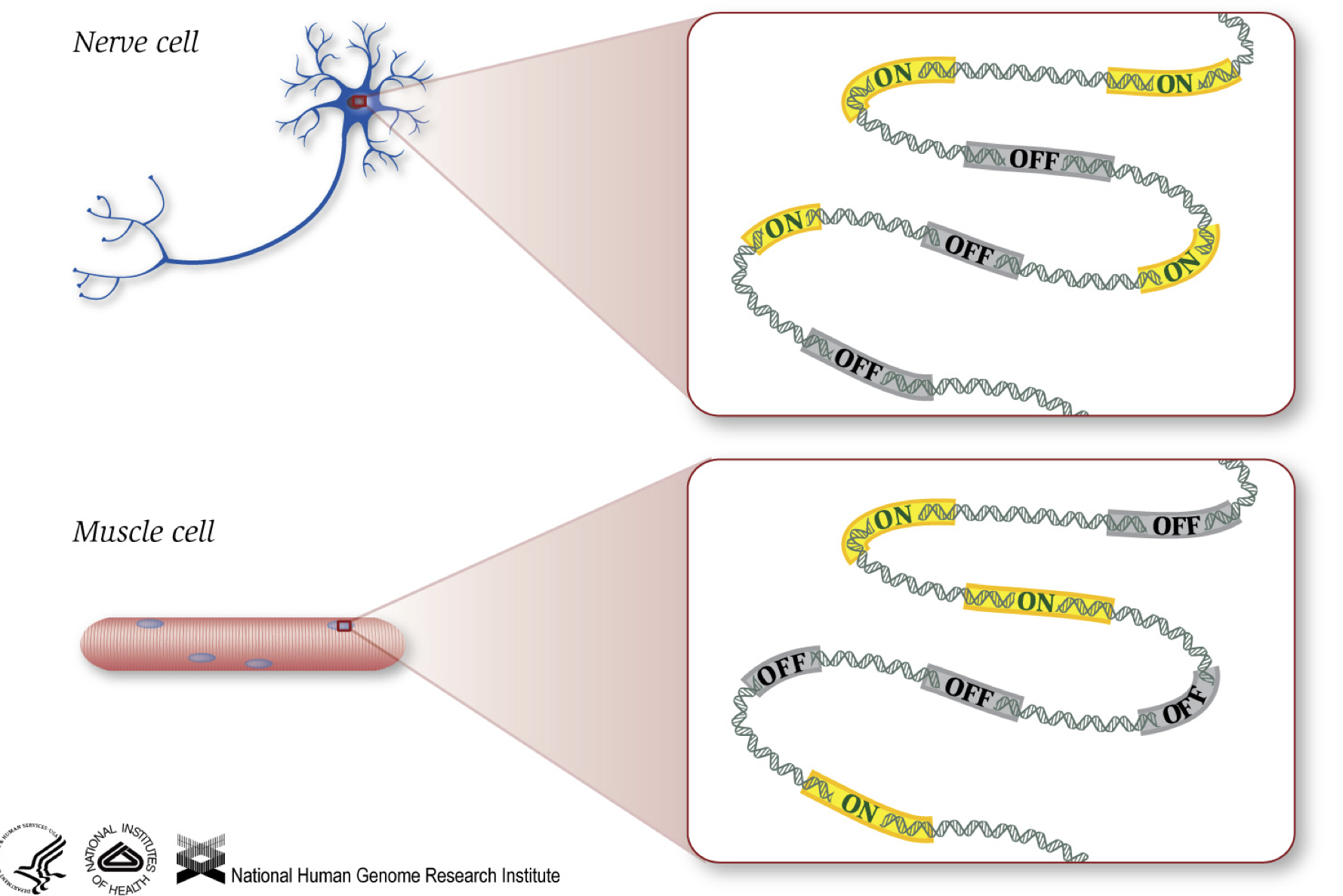
Erilaisia biologisia prosesseja, sekä mutaatioita, voivat vaikuttaa siihen, mitkä geenit ovat päällä ja jotka ovat pois päältä, lisäksi, kuinka paljon geenit ovat päällä/pois.,
Jotta proteiinit, DNA on puhtaaksi messenger RNA tai mRNA: ta, joka on käännetty ribosomin proteiineja. Kuitenkin, jotkut geenit koodaavat RNA: ta, että ei saada käännetty proteiini; nämä RNAs kutsutaan non-coding RNAs, tai ncRNAs. Usein nämä RNAs on toiminto ja itsestään ja ovat rRNAs, tRNAs, ja siRNAs, mm. Kaikkia geeneistä transkriptoituja RNAs-arvoja kutsutaan transkripteiksi.
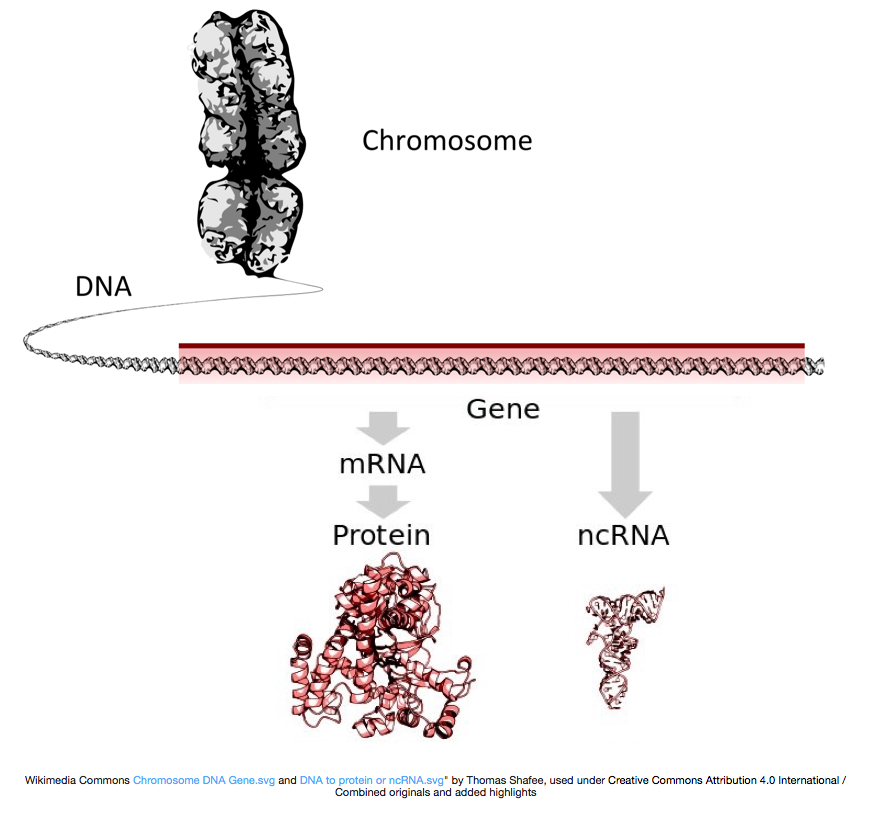
käännetty proteiineja, RNA on tehtävä käsittely tuottaa mRNA: ta., Alla olevassa kuvassa, päälisäie kuva edustaa geenin DNA: n, joka koostuu untranslated regions (UTRs) ja avaa lue runko. Geenit transkriboidaan pre-mRNA: ksi, joka sisältää edelleen intronisia sekvenssejä. Kun post-transciptional käsittely, intronit saumattu ulos ja polyA-hännän ja 5′ – cap lisätään tuotto kypsä mRNA selostukset, jotka voivat olla käännetty proteiineja.,
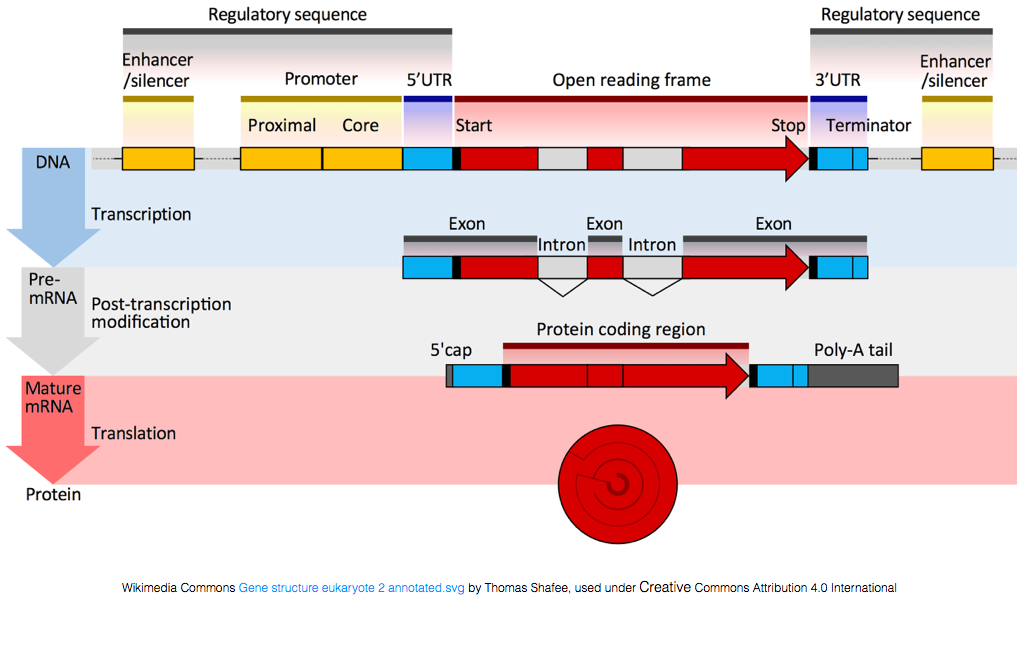
Kun mRNA selostukset on polyA-hännän, monet ei-koodaava RNA selostukset eivät ole niin post-transkription käsittely on eri näistä selostukset.
Transcriptomics
transcriptome on määritelty kokoelma kaikki transkriptio lukemat läsnä solussa., RNA-seq data voidaan tutkia ja/tai arvioida transcriptome organismin, joka voidaan käyttää seuraavan tyyppisiä kokeiluja:
- Ero Geenien Ilmentyminen: määrällinen arviointi ja vertailu transkriptio tasoilla
- Transcriptome kokoonpano: rakennuksen profiilia puhtaaksi alueita genomin, laadullinen arviointi.,
- Voidaan käyttää auttaa rakentamaan parempi geeni malleja, ja tarkistaa niitä käyttämällä kokoonpano
- Metatranscriptomics tai yhteisön transcriptome analyysi
Illumina library preparation
Kun alkaa RNA-seq-kokeen, joka näyte RNA on eristettävä ja kääntyi cDNA-kirjaston sekvensointi. Kirjaston valmistelun yleinen työnkulku on esitetty alla olevissa vaiheittaisissa kuvissa.
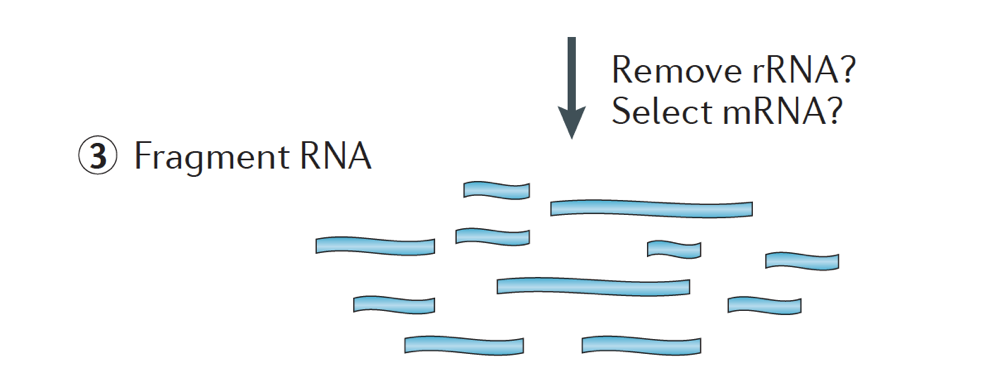
lyhyesti RNA eristetään näytteestä ja kontaminoiva DNA poistetaan Dnasella.,
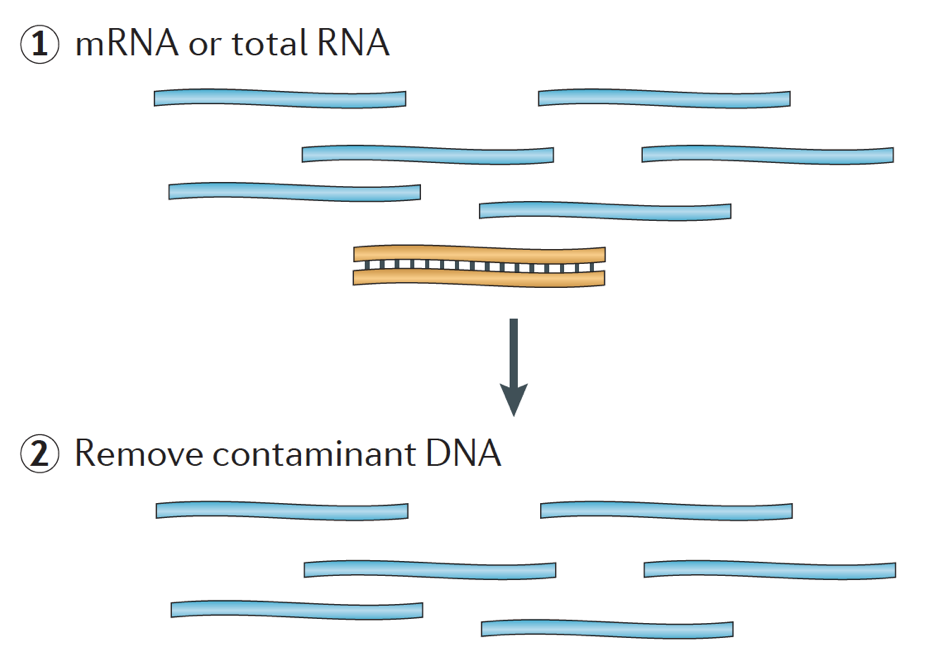
RNA-näyte sitten läpi joko valinta mRNA (polyA valinta) tai ehtyminen rRNA. Tuloksena oleva RNA on pirstaloitunut.
Yleensä, ribosomaalisen RNA: ta edustaa suurin osa RNAs läsnä solussa, kun messenger RNAs edustaa pieni prosenttiosuus total RNA, ~2% ihmisillä. Siksi, jos haluamme tutkia proteiinia koodaavan geenien, meidän täytyy rikastuttaa varten mRNA tai heikentävistä rRNA., Differential geenien ilmentyminen analyysi, se on parasta palvelua ja kaikki olennaiset Poly(A)+, jos olet tavoitteena saada tietoa pitkän non-coding RNAs, sitten tehdä ribosomaalisen RNA: n ehtyminen.
koko kohde palasia lopullisessa kirjasto on keskeinen parametri kirjaston rakentaminen. DNA: n pirstoutuminen tapahtuu tyypillisesti fysikaalisilla menetelmillä (eli akustisella leikkauksella ja sonikaatiolla) tai entsymaattisilla menetelmillä (eli epäspesifisillä endonukleaasicocktaileilla ja transposaasi-tagmentaatioreaktioilla.,
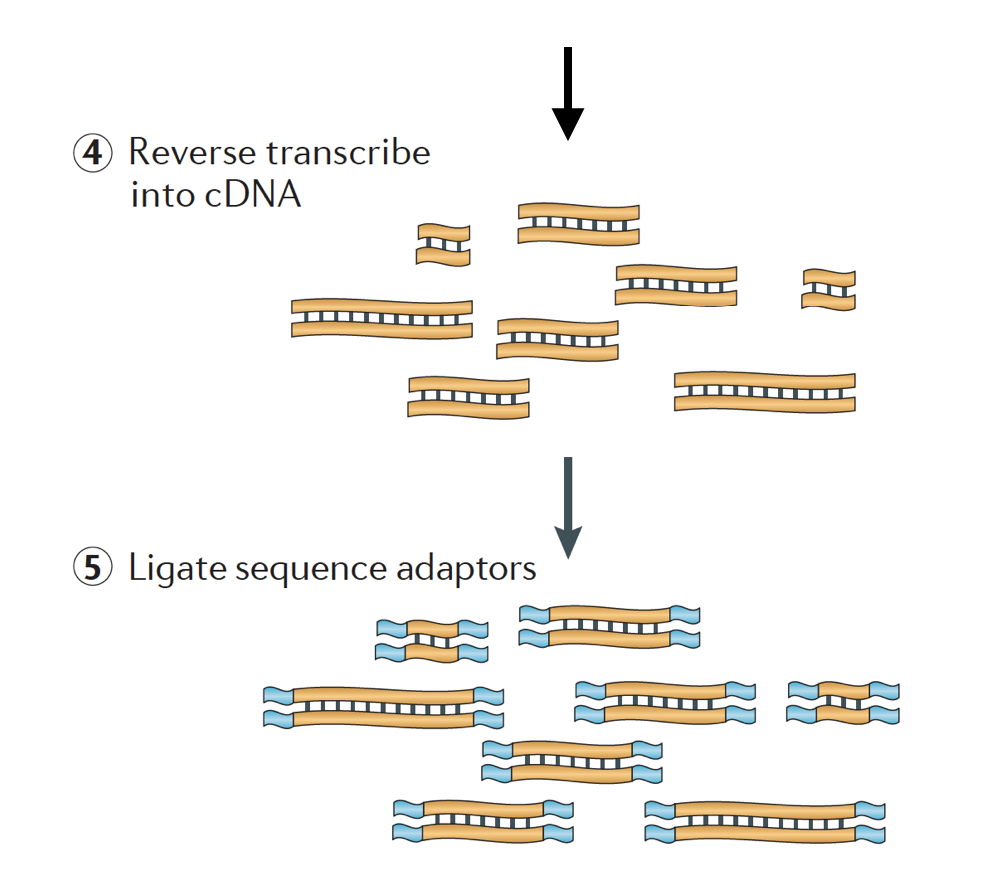
RNA on sitten käänteinen puhtaaksi osaksi kaksijuosteista cDNA ja järjestyksessä sovittimet ovat sitten lisätään päät palasia.
cDNA-kirjastoja voidaan luoda tapa säilyttää tietoa siitä, mikä osa DNA-RNA oli transkriptoitu. Kirjastot, jotka säilyttävät tämän tiedon, ovat nimeltään stranded kirjastot, jotka ovat nyt vakiona Illumina ’ s TruSeq stranded RNA-Seq sarjat., Jumiutuneet kirjastot eivät saisi olla sen kalliimpia kuin kahlitsemattomat, joten ei ole oikeastaan mitään syytä olla hankkimatta tätä lisätietoa.,
On olemassa 3 eri cDNA-kirjastoista saatavilla:
- Eteenpäin (secondstrand) – lukee muistuttavat geenin sekvenssi tai secondstrand cDNA-sekvenssi
- Käänteinen (firststrand) – lukee muistuttavat täydentää geenin sekvenssi tai firststrand cDNA-sekvenssi (TruSeq)
- Unstranded
Lopuksi, fragmentit PCR-monistetaan, jos tarvitaan, ja fragmentit ovat kooltaan valittu (yleensä ~300-500bp) loppuun library.
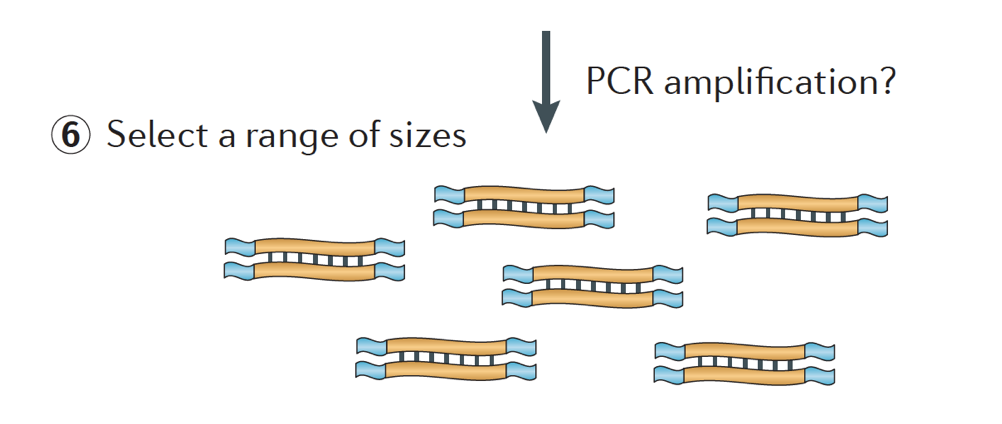
Kuva luotto: Martin, J. A., Wang, Z., Nat. Ilm., Genet. (2011) 12:671-682
Illumina Sekvensointi
Single-end vs. Pariksi-end
sen Jälkeen, kun valmistelu kirjastot, sekvensointi voidaan suorittaa tuottaa nukleotidisekvenssit päät fragmentteja, jotka ovat nimeltään lukee. Sinulla on valinta sekvensointi yhden vuoden cDNA-fragmentteja (single-end lukee) tai molemmat päät fragmentteja (pariksi-end lukee).,
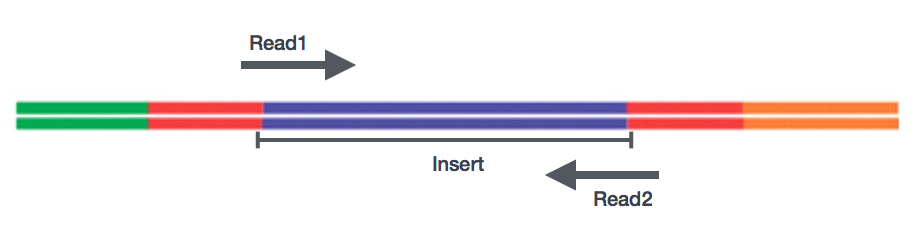
- SE – Single end dataset => Vain Read1
- PE – Pariksi-end-aineisto => Read1 + Read2
- voi olla 2 erillistä FastQ tiedostoja, tai vain yksi limitetään pareittain
Yleisesti single-end-sekvensointi on riittävä, ellei se ole odotettavissa, että lukee täsmää useita sijainteja genomin (esim. organismien kanssa monet paralogous geenit), kokoonpanot suoritetaan, tai liitos-isoentsyymin eriyttäminen. Huomaa, että pariläpäiset lukemat ovat yleensä 2x kalliimpia.,
Eri sekvensointi alustoilla
On olemassa erilaisia Illumina alustoja valita sekvenssin ekspressio kirjastot.

Kuva luotto: Mukailtu Illumina
Erot foorumi voi muuttaa pituus lukee syntyy, laatu lukee, sekä kokonaismäärä lukee sekvensoitiin per juosta ja tarvittava aika järjestyksessä kirjastot., Eri alustoilla, jokainen käyttää eri virtaus solun, joka on lasin pinta on päällystetty järjestely pariksi oligos, jotka täydentävät adapterit lisätään malliin molekyylejä. Virtaussolussa sekvensointireaktiot tapahtuvat.

Kuva luotto: Mukailtu Illumina
Sekvensointi-by-synteesi
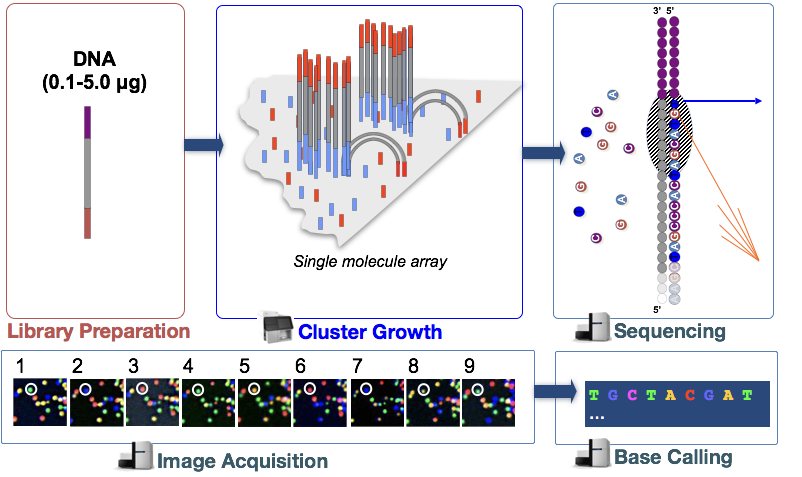
Illumina sekvensointi teknologia käyttää sekvensointi-by-synthesis lähestymistapa, jota on kuvattu tarkemmin alla.
vaiheessa cDNA-kirjaston DNA-fragmentit denaturoidaan ja levitetään lasivirtauskennoon., Nämä denaturoitu fragmentit sitoutuvat täydentäviä oligos, jotka ovat jo sitoutuneet kovalenttisesti virtaus solun kaistaa, jolloin kiinnitys.
Klusterin Sukupolven
Kun palaset on kiinnitetty, vaihe, nimeltään klusterin sukupolvi alkaa. Tämän vaiheen aikana yksittäisiä fragmentteja monistetaan klonaalisesti siten, että syntyy identtisten fragmenttien klusteri (fragmentteja lähellä). Tämä on välttämätöntä, jotta fluoresenssi voidaan helposti jää jokaisen klusterin, sen sijaan yhden fragmentti aikana nukleotidin sisällyttämistä seuraavaan vaiheeseen.,
- Koota täydentää polymeraasi
- dsDNA on denaturoitu, ja alkuperäinen DNA pestään pois jättäen syntetisoitu juoste kovalenttisesti sitoutuneet flow cell.
- Yhden lohkon hybridises, jonka vieressä sovitin muodostaa ”sillan”
- dsDNA on laajennettu-polymeraasi. Jokainen nauha kovalenttisesti sidottu eri adapteri.
- toistaa monta kertaa, jotta klonaalisesti vahvistuvat kaikki virtaussolun ainutlaatuiset fragmentit muodostaen identtisen sekvenssin klustereita.,
Sequencing by synthesis (& image acquisition)
sen Jälkeen, kun klusterin sukupolvi, fluoresoivasti-tagged nukleotidit on otettu yksi kerrallaan (syklisesti) ja fluoresenssi kuvat ovat kaapattu mitkä nukleotidin saa osaksi kunkin klusterin jokaisen syklin aikana.
- Denaturoida klustereita ja lohko 3′ päät estää ei-toivotut pohjustus.
- Hybridisoitua sekvensointi alukkeiden adapter sekvenssi lankoja.,
- sykli neljä NTPs kanssa fluoresoiva markkereita ja terminator sekvenssi ja polymeraaseja.
- Kun NTP on otettu, klusteri on innoissaan valonlähde ja ominaisuus fluroscent signaali lähetetään.
- väri kirjataan, sitten väriaineen terminaattori halkaistaan ja pestään. Prosessi toistaa tietyn määrän syklejä.,
Pohja Soittamalla
Illumina on oma ohjelmisto, joka käy läpi kaikki otetut kuvat edellisessä vaiheessa ja tuottaa tekstitiedostoja järjestyksessä tiedot jokaisen klusterin, joka perustuu fluoresenssi. Lisäksi soittamalla emäkset, tämä ohjelmisto määrittää probablity pisteet osoittavat, miten tietyt se oli, että kutsuu jotain ”A”, ”T”, ”G” tai ”C”.
– Jos on epäselvyyksiä, esim., tietty sykli kuvaa klusterin ei ole erillisiä väri, joka voi liittyä tietyn nukleotidin pohja soittamalla ohjelmisto on pieni todennäköisyys liittyy siihen ja antaa ”N” eikä ”A”, ”T”, ”G” tai ”C”.
lopuksi,
- Useita klustereita ~= Määrä lukee
- Määrä sekvensointi sykliä = Pituus lukee
useita sykliä (pituus lukee) riippuu sekvensointi alusta käytetään sekä mieltymykset.
HUOM., Jos haluat tutkia sekvensointia synteesin avulla syvällisemmin, suosittelemme tätä todella mukavaa animaatiota, joka on saatavilla Illuminan YouTube-kanavalla.
Multiplexing
Riippuen Illumina alusta (MiSeq, HiSeq, NextSeq), kaistojen lukumäärä per virtaus solun, ja määrä lukee, että voidaan saada per kaista vaihtelee suuresti. Sinun täytyy päättää, kuinka monta lukee haluaisit per näyte (eli sequencning syvyys) ja sitten perustuu alustan voit valita, laskea, kuinka monta kaistaa tarvitaan oman joukko näytteitä., Me puhua enemmän näkökohdat tehdessään tätä päätöstä seuraavan oppitunnin Kokeellisen Näkökohtia,
Tyypillisesti, maksut sekvensointi ovat kaistaa kohti flow cell ja voit ajaa useita näytteitä per kaista. Illumina onkin kehittänyt mukavan multiplexing-menetelmän, jonka avulla useiden näytteiden kirjastot voidaan yhdistää ja sekvensoida samanaikaisesti virtaussolun samalla kaistalla. Tämä menetelmä edellyttää indeksien (Illumina-sovittimen sisällä) tai erityisten viivakoodien (Illumina-sovittimen ulkopuolella) lisäämistä alla olevassa kaaviossa kuvatulla tavalla.,
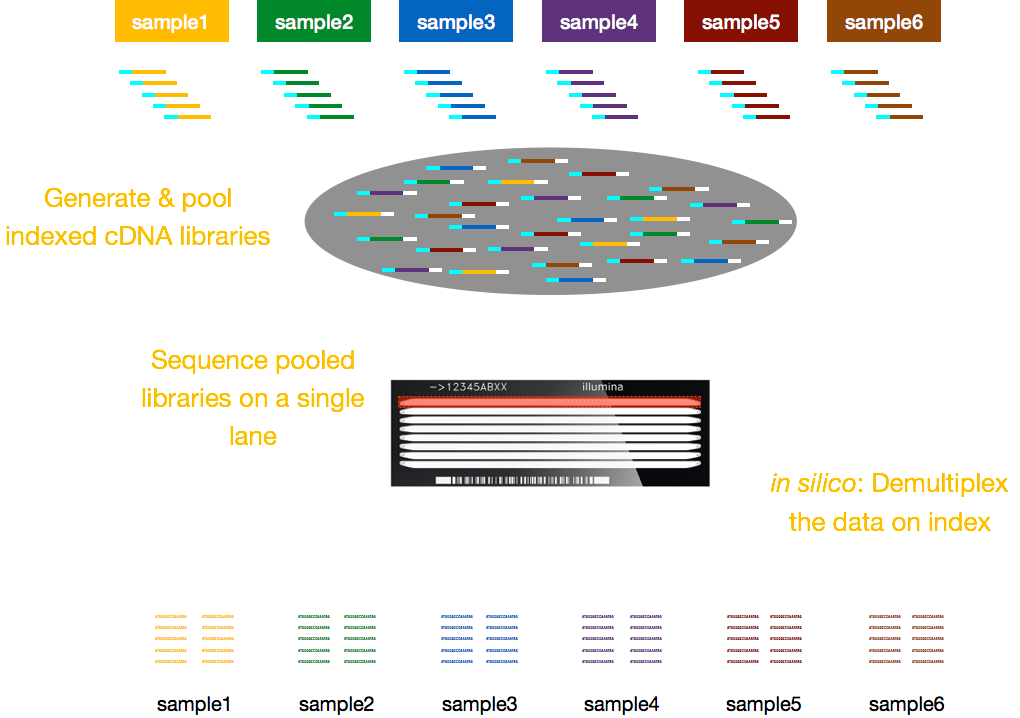
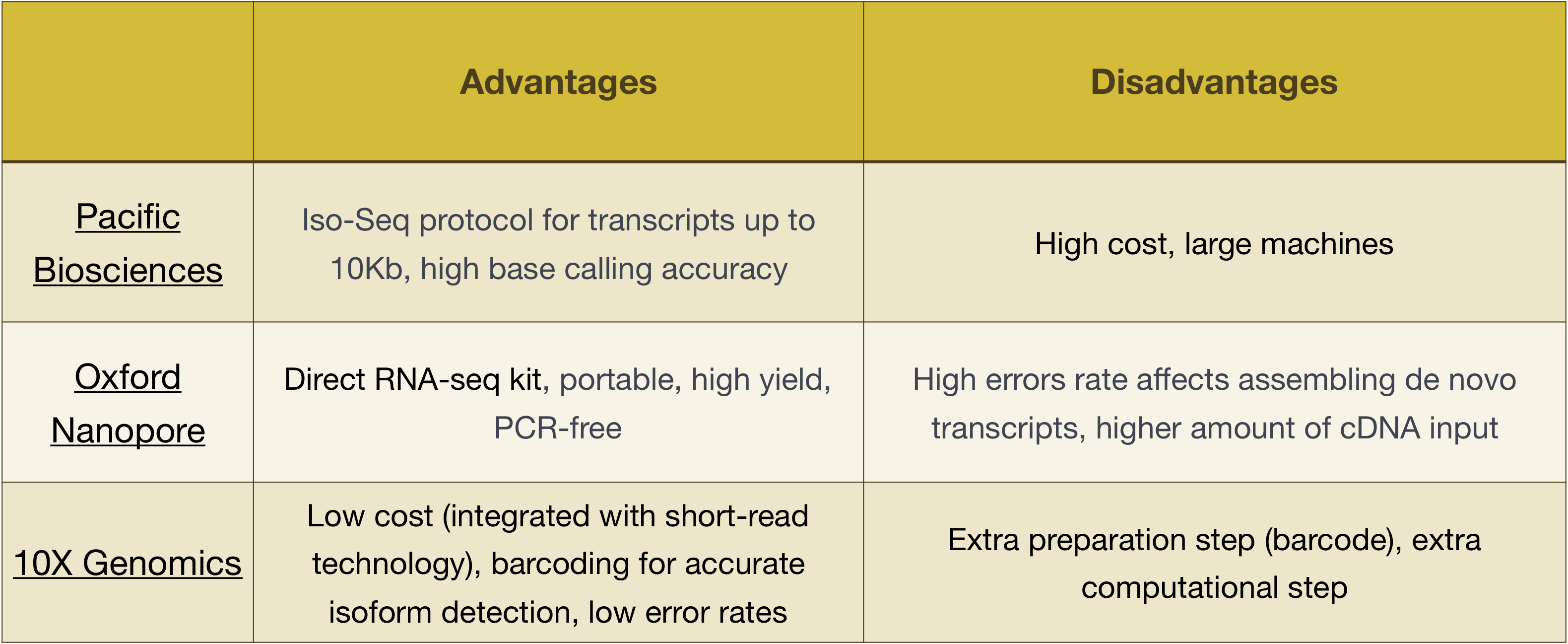
HUOMAUTUS: työnkulku on esitetty tämä opetus on nimenomaan Illumina sekvensointi, joka on tällä hetkellä eniten käytetty menetelmä sekvensointi., Mutta on olemassa muita pitkä-lue sekvensointi menetelmät syytä huomata, kuten:
- Tyynenmeren Biosciences: http://www.pacb.com/
- Oxford Nano (MinION): https://nanoporetech.com/
- 10X Genomiikka: https://www.10xgenomics.com/
Edut ja haitat näitä tekniikoita voidaan tarkastella seuraavassa taulukossa:
Vastaa