temps approximatif: 90 minutes
objectifs D’apprentissage:
- décrire le processus de préparation de la bibliothèque RNA-seq
- décrire la méthode de séquençage Illumina
technique expérimentale passionnante qui est utilisée pour explorer et/ou quantifier l’expression des gènes dans ou entre les conditions.,
Comme nous le savons, les gènes fournissent des instructions pour fabriquer des protéines, qui remplissent certaines fonctions au sein de la cellule. Bien que toutes les cellules contiennent la même séquence D’ADN, les cellules musculaires sont différentes des cellules nerveuses et d’autres types de cellules en raison des différents gènes qui sont activés dans ces cellules et des différents ARN et protéines produits.
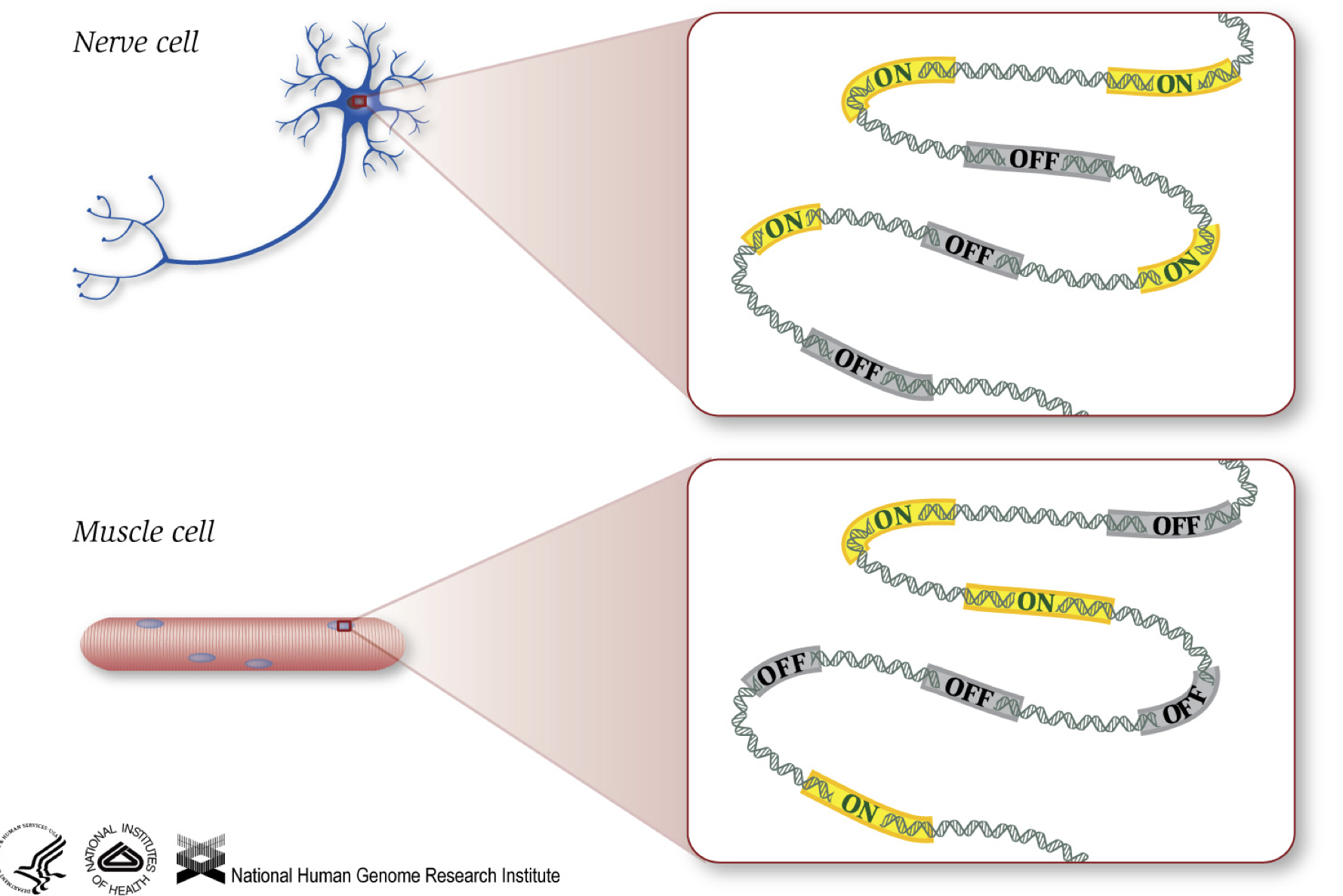
les Différents processus biologiques, ainsi que des mutations peuvent affecter dont les gènes sont activés et qui sont éteints, en outre, combien de gènes spécifiques sont allumé/éteint.,
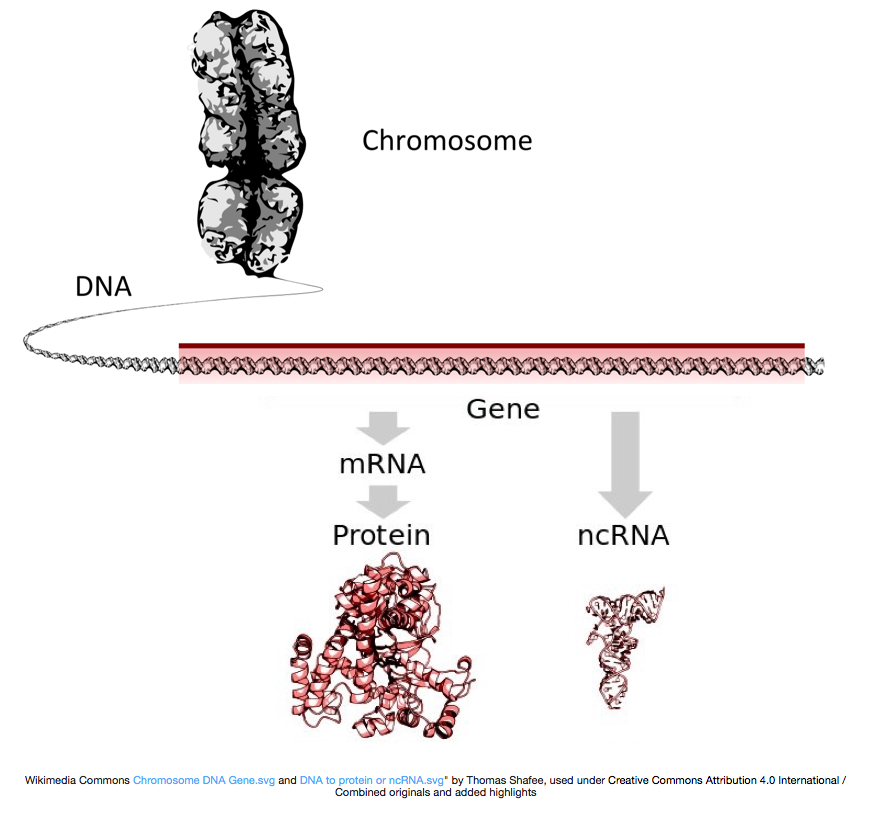
pour fabriquer des protéines, l’ADN est transcrit en ARN messager, ou ARNm, qui est traduit par le ribosome en protéine. Cependant, certains gènes codent L’ARN qui ne se traduit pas en protéine; ces ARN sont appelés ARN non codants, ou ncRNAs. Souvent, ces ARN ont une fonction en eux-mêmes et comprennent les ARNr, les ARNt et les ARNr, entre autres. Tous les ARN transcrits à partir de gènes sont appelés transcriptions.
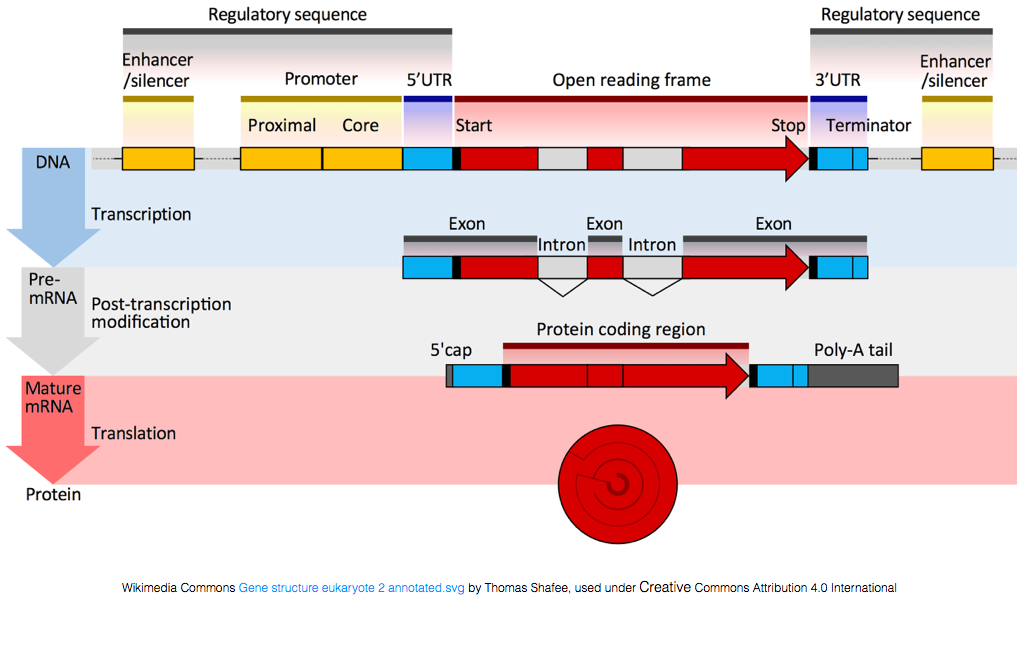
Pour être traduit en protéines, l’ARN doit subir un traitement pour générer l’ARNm., Dans la figure ci-dessous, le brin supérieur de l’image représente un gène dans L’ADN, composé des régions non traduites (UTR) et du cadre de lecture ouvert. Les gènes sont transcrits en pré-ARNm, qui contient toujours les séquences introniques. Après le traitement post-transciptionnel, les introns sont épissés et une queue de polyA et un capuchon de 5’ sont ajoutés pour produire des transcriptions d’ARNm matures, qui peuvent être traduites en protéines.,
bien que les transcriptions d’ARNm aient une queue polyA, la plupart des transcriptions D’ARN non codantes ne le font pas car le traitement post-transcriptionnel est différent pour ces transcriptions.
transcriptomique
le transcriptome est défini comme un ensemble de toutes les lectures de transcription présentes dans une cellule., Les données ARN-seq peuvent être utilisées pour explorer et/ou quantifier le transcriptome d’un organisme, qui peut être utilisé pour les types d’expériences suivantes:
- Expression génique différentielle: évaluation quantitative et comparaison des niveaux de transcription
- assemblage du Transcriptome: construction du profil des régions transcrites du génome, une évaluation qualitative.,
- peut être utilisé pour aider à construire de meilleurs modèles de gènes et les vérifier à l’aide de L’assemblage
- Métatranscriptomique ou Analyse du transcriptome communautaire
préparation de la bibliothèque Illumina
lors du démarrage d’une expérience ARN-seq, pour chaque échantillon, l’ARN doit être isolé et transformé en Le flux de travail général pour la préparation de la Bibliothèque est détaillé dans les images étape par étape ci-dessous.
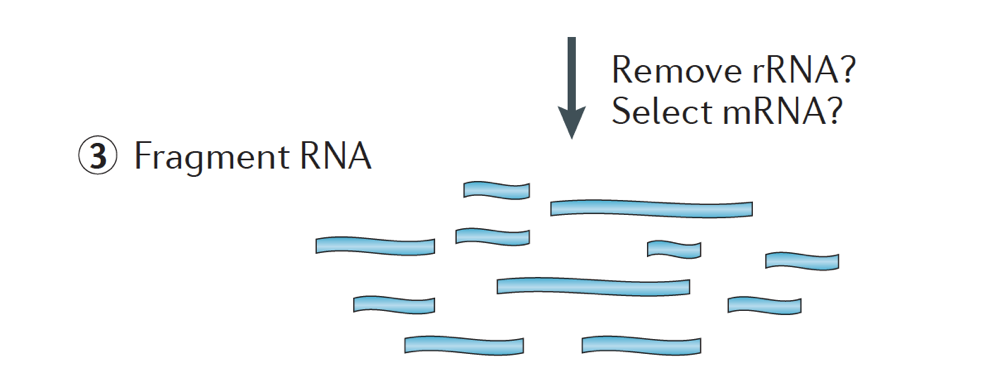
brièvement, l’ARN est isolé de l’échantillon et l’ADN contaminant est éliminé avec la DNase.,
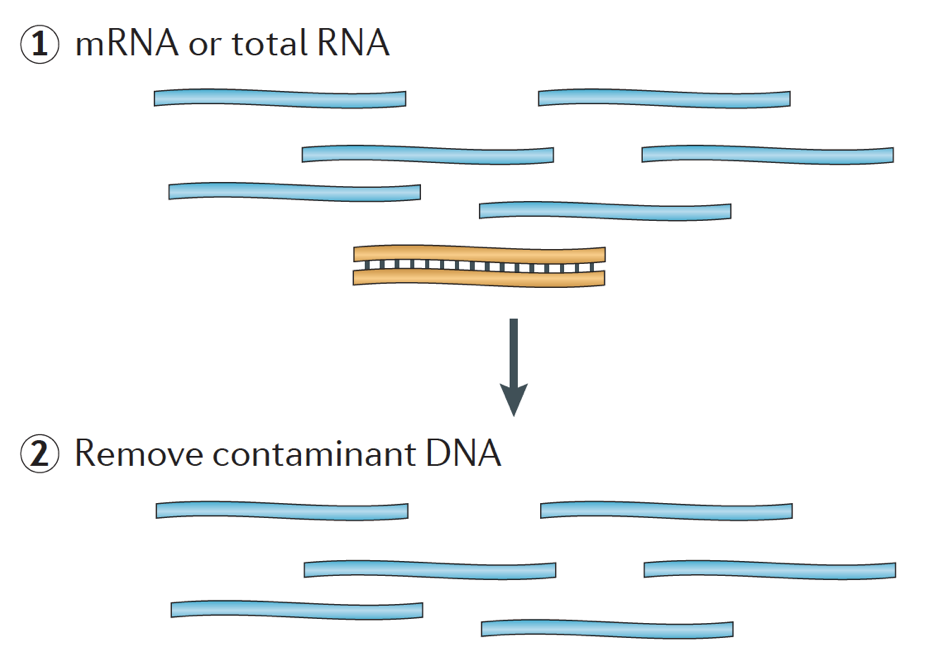
l’échantillon D’ARN subit alors soit une sélection de l’ARNm (sélection polyA), soit une déplétion de l’ARNr. L’ARN résultant est fragmenté.
En Général, L’ARN ribosomique représente la majorité des ARN présents dans une cellule, tandis que les ARN messagers représentent un faible pourcentage de L’ARN total, ~2% chez l’homme. Par conséquent, si nous voulons étudier les gènes codant les protéines, nous devons enrichir l’ARNm ou épuiser l’ARNr., Pour l’analyse différentielle de l’expression des gènes, il est préférable d’enrichir pour Poly(A)+, à moins que vous ne visiez à obtenir des informations sur les ARN longs non codants, puis de faire une déplétion de L’ARN ribosomique.
la taille des fragments cibles dans la bibliothèque finale est un paramètre clé pour la construction de la bibliothèque. La fragmentation de l’ADN se fait généralement par des méthodes physiques (cisaillement acoustique et sonication) ou enzymatiques (cocktails d’endonucléases non spécifiques et réactions de tagmentation de transposase.,
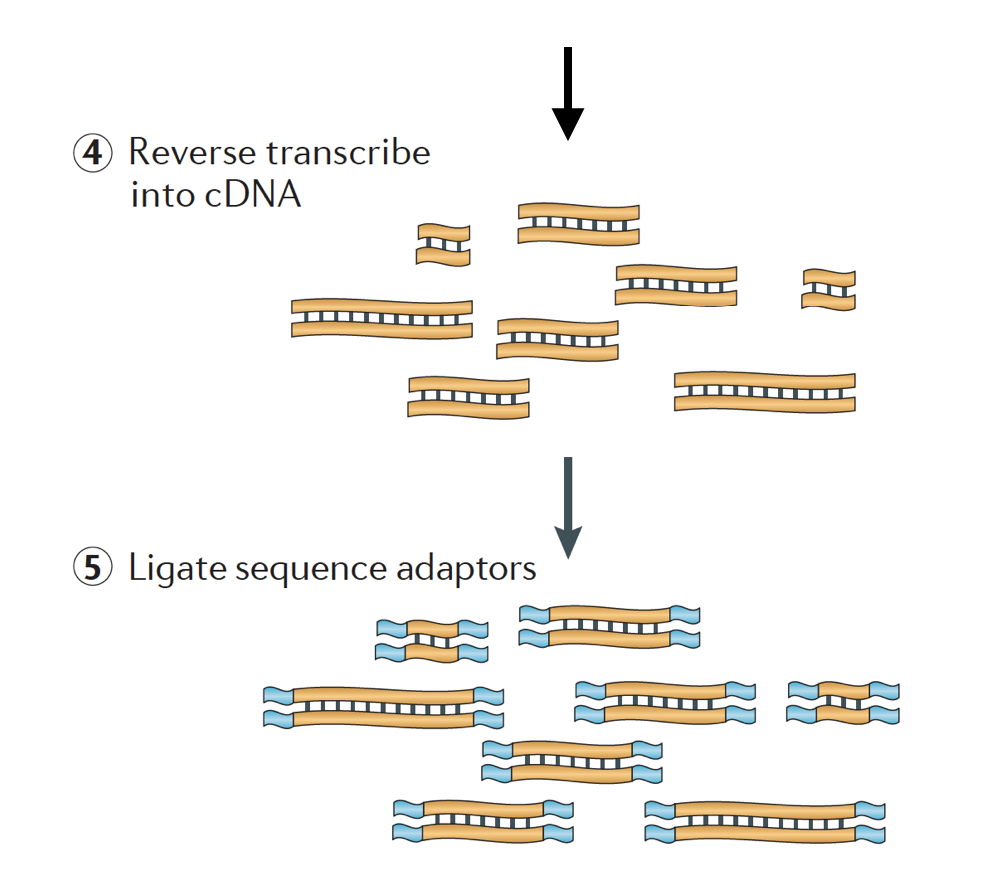
L’ARN est ensuite transcrit en inverse double brin d’adnc et de la séquence de cartes sont ensuite ajoutés aux extrémités des fragments.
Les bibliothèques d’ADNc peuvent être générées de manière à conserver des informations sur le brin d’ADN à partir duquel l’ARN a été transcrit. Les bibliothèques qui conservent ces informations sont appelées bibliothèques échouées, qui sont maintenant standard avec les kits TruSeq stranded ARN-Seq D’Illumina., Les bibliothèques bloquées ne devraient pas être plus chères que les bibliothèques non marquées, il n’y a donc pas vraiment de raison de ne pas acquérir ces informations supplémentaires.,
il existe 3 types de bibliothèques d’ADNc disponibles:
- Forward (secondstrand) – les lectures ressemblent à la séquence du gène ou à la séquence de l’ADNc secondstrand
- Reverse (firststrand) – les lectures ressemblent au complément de la séquence du gène ou de la séquence de l’ADNc firststrand (TruSeq)
- Sans marque
enfin, les fragments sont amplifiés par PCR si nécessaire, et la taille des fragments est sélectionnée (généralement ~300-500bp) pour terminer la bibliothèque.
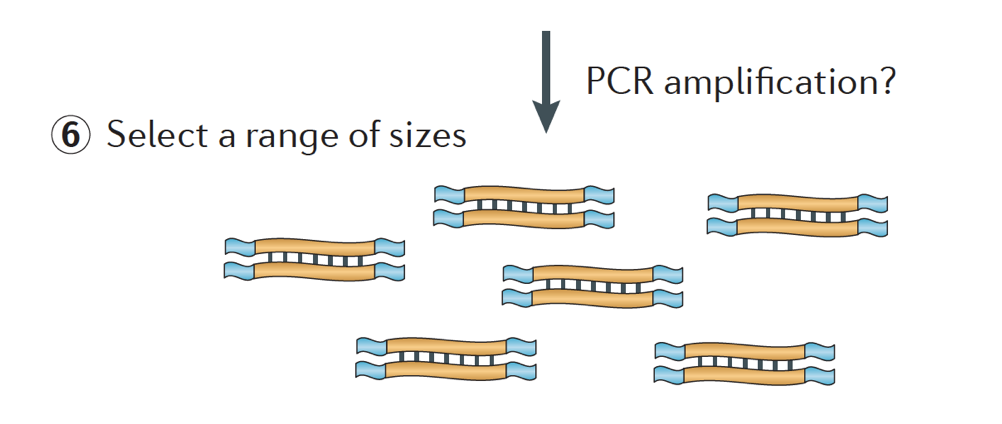
crédit D’Image: Martin J. A. et Wang Z., Nat. Rév., Genet. (2011) 12:671-682
séquençage Illumina
Single-end versus Paired-end
Après préparation des bibliothèques, le séquençage peut être effectué pour générer les séquences nucléotidiques des extrémités des fragments, appelées lectures. Vous aurez le choix de séquencer une seule extrémité des fragments d’ADNc (lectures à extrémité unique) ou les deux extrémités des fragments (lectures à extrémité appariée).,
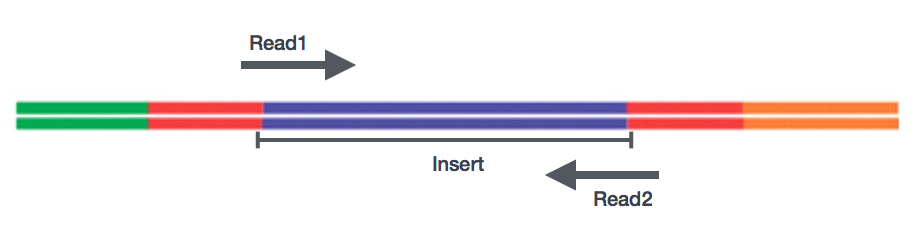
- Se – single end dataset => seulement Read1
- PE – Paired-end dataset => Read1 + Read2
- peut être 2 séparé fichiers FASTQ ou un seul avec des paires entrelacées
généralement, le séquençage à une extrémité est suffisant, sauf S’il est prévu que les lectures correspondent à plusieurs emplacements sur le génome (par exemple, des organismes avec de nombreux gènes paralogues), des assemblages sont effectués ou pour la différenciation des isoformes d’épissure. Sachez que les lectures appariées sont généralement 2 fois plus chères.,
différentes plates-formes de séquençage
Il existe une variété de plates-formes Illumina parmi lesquelles choisir pour séquencer les bibliothèques d’ADNc.

crédit D’Image: adapté D’Illumina
Les différences de plate-forme peuvent modifier la longueur des lectures générées, la qualité des lectures, ainsi que le nombre total de lectures séquencées par exécution et le temps nécessaire pour séquencer les bibliothèques., Les différentes plates-formes utilisent chacune une cellule d’écoulement différente, qui est une surface en verre revêtue d’un agencement d’oligos appariés qui sont complémentaires aux adaptateurs ajoutés à vos molécules de gabarit. La cellule d’écoulement est où le séquençage de réactions.

crédit D’Image: adapté D’Illumina
séquençage par synthèse
La technologie de séquençage Illumina utilise une approche de séquençage par synthèse qui est décrite plus en détail ci-dessous.
dans l’étape, les fragments D’ADN dans la bibliothèque d’ADNc sont dénaturés et appliqués à la cellule de flux de verre., Ces fragments dénaturés se lient aux oligos complémentaires qui sont déjà liés de manière covalente aux voies cellulaires d’écoulement, ce qui entraîne une fixation.
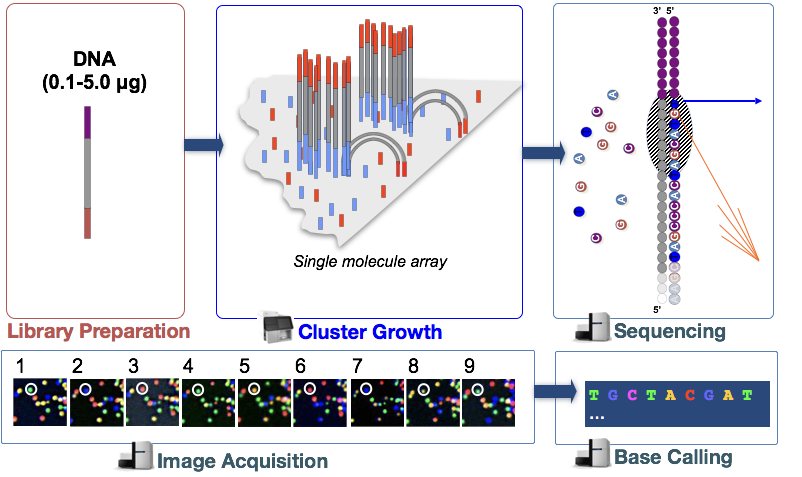
Cluster Génération
une Fois les fragments ont fixé, une phase appelée cluster génération commence. Au cours de cette étape, des fragments uniques sont amplifiés clonalement pour créer un cluster (fragments à proximité) de fragments identiques. Ceci est nécessaire pour que la fluorescence puisse être facilement capturée à partir de chaque cluster, au lieu d’un seul fragment, lors de l’incorporation de nucléotides à l’étape suivante.,
- synthétiser le complément avec la polymérase
- l’ADN dsDNA est dénaturé et l’ADN original emporté laissant le brin synthétisé lié de manière covalente à la cellule d’écoulement.
- Un seul brin s’hybride avec un adaptateur adjacent pour former un « pont »
- l’adnsd est prolongé par polymérase. Chaque brin lié de manière covalente à un adaptateur différent.
- répétez plusieurs fois pour amplifier cloniquement tous les fragments uniques sur la cellule de flux pour former des grappes de séquence identique.,
séquençage par synthèse (& acquisition d’images)
Après la génération de grappes, des nucléotides marqués par fluorescence sont incorporés un à la fois (cycliquement) et des images de fluorescence sont capturées pour identifier quel nucléotide est incorporé dans chaque grappe
- dénaturer les clusters et les extrémités du bloc 3’ pour éviter un amorçage indésirable.
- hybrider les amorces de séquençage à la séquence de l’adaptateur aux extrémités libres.,
- Cycle quatre NTP avec marqueurs fluorescents et séquence de terminaison et polymérases.
- Une fois NTP incorporé, le cluster est excité par une source lumineuse et un signal fluroscent caractéristique est émis.
- La couleur est enregistré, puis le terminateur sur le colorant est clivé et lavé. Répéter le processus pour un nombre de cycles précis.,
appel de Base
Illumina dispose d’un logiciel propriétaire qui parcourt toutes les images capturées à l’étape précédente et génère des fichiers texte avec des informations de séquence sur chaque cluster en fonction de la fluorescence. En plus d’appeler les bases, ce logiciel attribue un score de probabilité pour indiquer à quel point il était certain de l’appel d’un « A”, d’un « T”, d’un « G” ou d’un « C”.
S’il y a des ambiguïtés, par exemple, à un certain cycle, l’image d’un cluster n’a pas de couleur distincte pouvant être associée à un nucléotide spécifique, le logiciel d’appel de base aura une faible probabilité associée et attribuera un « N” au lieu de « A”, « T”, « G” ou « C”.
En conclusion,
- Nombre de clusters ~= Nombre de lit
- Nombre de cycles de séquençage = Longueur du lit
Le nombre de cycles (longueur du lit) dépend de la plate-forme de séquençage utilisée ainsi que de vos préférences.
REMARQUE., Si vous souhaitez explorer le séquençage par synthèse plus en profondeur, nous vous recommandons cette très belle animation disponible sur la chaîne Youtube D’Illumina.
multiplexage
selon la plate-forme Illumina (MiSeq, HiSeq, NextSeq), le nombre de voies par cellule de flux et le nombre de lectures pouvant être obtenues par voie varient considérablement. Vous devrez décider du nombre de lectures que vous souhaitez par échantillon (c’est-à-dire la profondeur de séquençage), puis en fonction de la plate-forme que vous choisissez, calculez le nombre total de voies dont vous aurez besoin pour votre ensemble d’échantillons., Nous parlerons plus de considérations lors de la prise de cette décision dans la prochaine leçon sur les considérations expérimentales
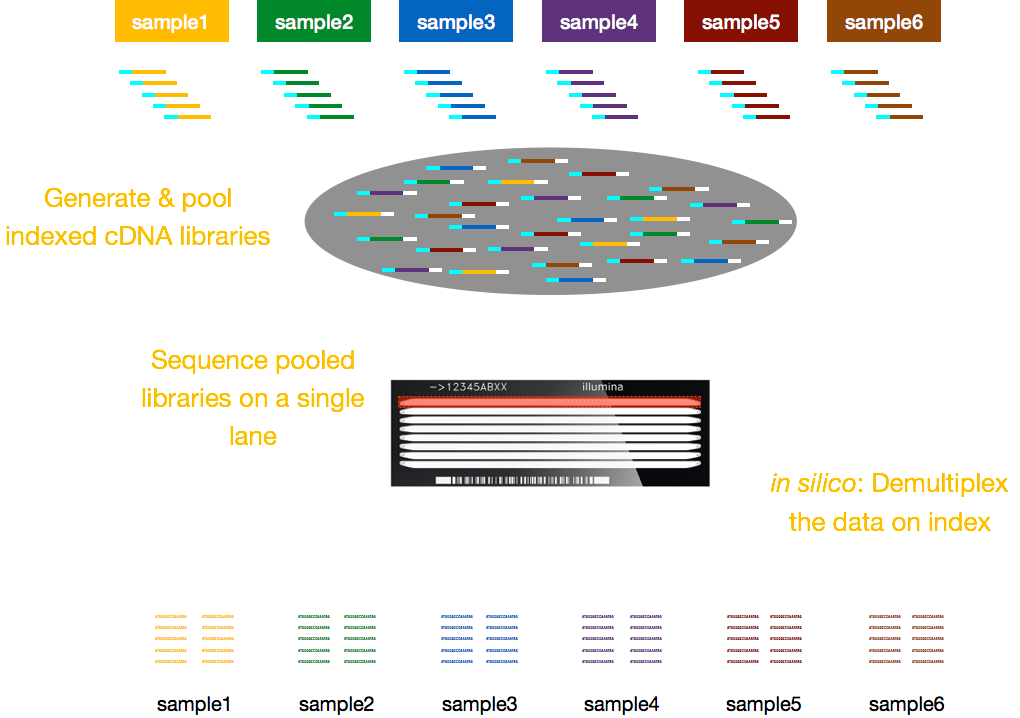
En règle générale, les frais de séquençage sont par voie de la cellule de flux et vous pourrez exécuter plusieurs échantillons par voie. Illumina a donc conçu une belle méthode de multiplexage qui permet de regrouper et de séquencer simultanément des bibliothèques de plusieurs échantillons dans la même voie d’une cellule de flux. Cette méthode nécessite l’ajout d’indices (à l’intérieur de L’adaptateur Illumina) ou de codes à barres spéciaux (à l’extérieur de l’adaptateur Illumina) comme décrit dans le schéma ci-dessous.,
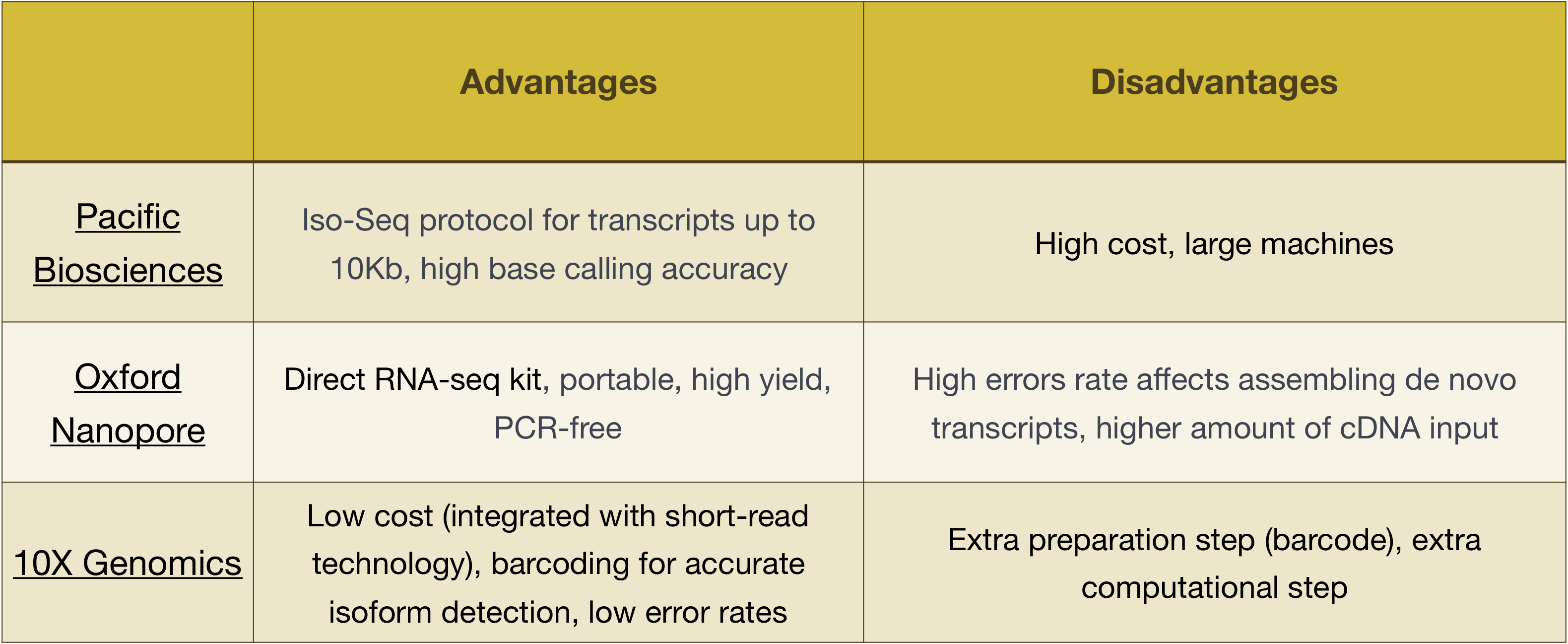
remarque: le flux de travail présenté dans cette leçon est spécifique au séquençage Illumina, qui est actuellement la méthode de séquençage la plus utilisée., Mais il y a d’autres long-lire des méthodes de séquençage à noter, tels que:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Génomique: https://www.10xgenomics.com/
les Avantages et les inconvénients de ces technologies peuvent être explorées dans le tableau ci-dessous:
Laisser un commentaire