Hozzávetőleges idő: 90 perc
Tanulási Célok:
- írja le a folyamat, hogy az RNS-seq könyvtár készítmény
- Leírni az Illumina szekvenálási módszer
Bevezetés RNS-seq
RNS-seq egy izgalmas kísérleti technika, hogy használni, hogy vizsgálja meg, és/vagy számszerűsíteni génexpresszió belül, illetve azok között a feltételek.,
mint tudjuk, a gének utasításokat adnak a fehérjék előállítására, amelyek bizonyos funkciókat látnak el a sejten belül. Bár minden sejt ugyanazt a DNS-szekvenciát tartalmazza, az izomsejtek különböznek az idegsejtektől és más típusú sejtektől az ezekben a sejtekben bekapcsolt különböző gének, valamint az előállított különböző RNS-ek és fehérjék miatt.
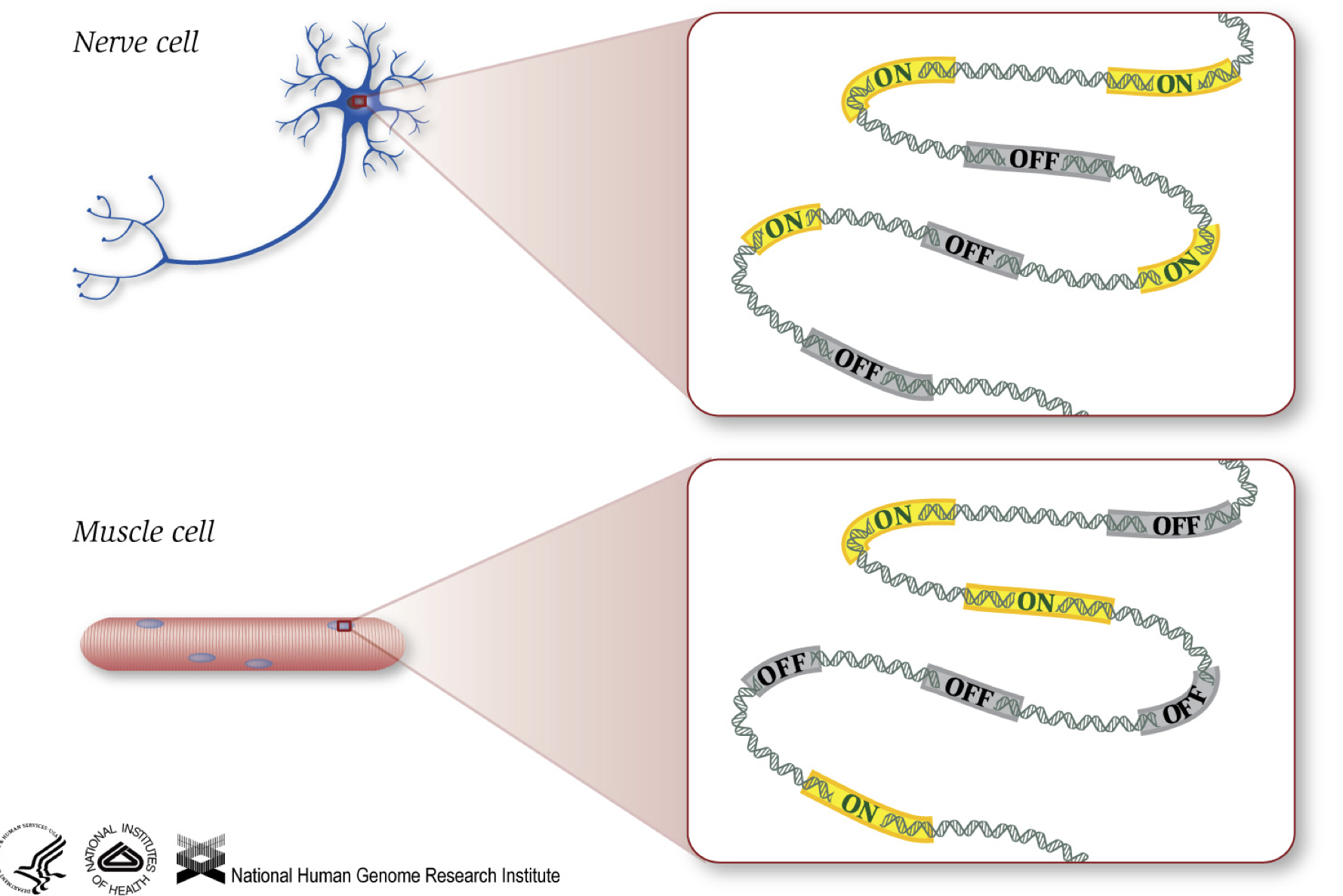
különböző biológiai folyamatok, valamint mutációk befolyásolhatják, hogy mely gének vannak bekapcsolva és melyek ki vannak kapcsolva, továbbá, hogy mennyi specifikus gének vannak bekapcsolva/kikapcsolva.,
a fehérjék előállításához a DNS-t messenger RNS-re vagy mRNS-re írják át, amelyet a riboszóma fehérjévé fordít. Azonban néhány gén kódol RNS, hogy nem lefordították fehérje; ezek az Rns nevezzük a nem-kódoló Rns, vagy ncRNAs. Ezek az RNS-ek gyakran önmagukban is működnek, többek között rrnas, tRNAs és siRNAs. A génekből átírt összes RNS-t átiratnak nevezik.
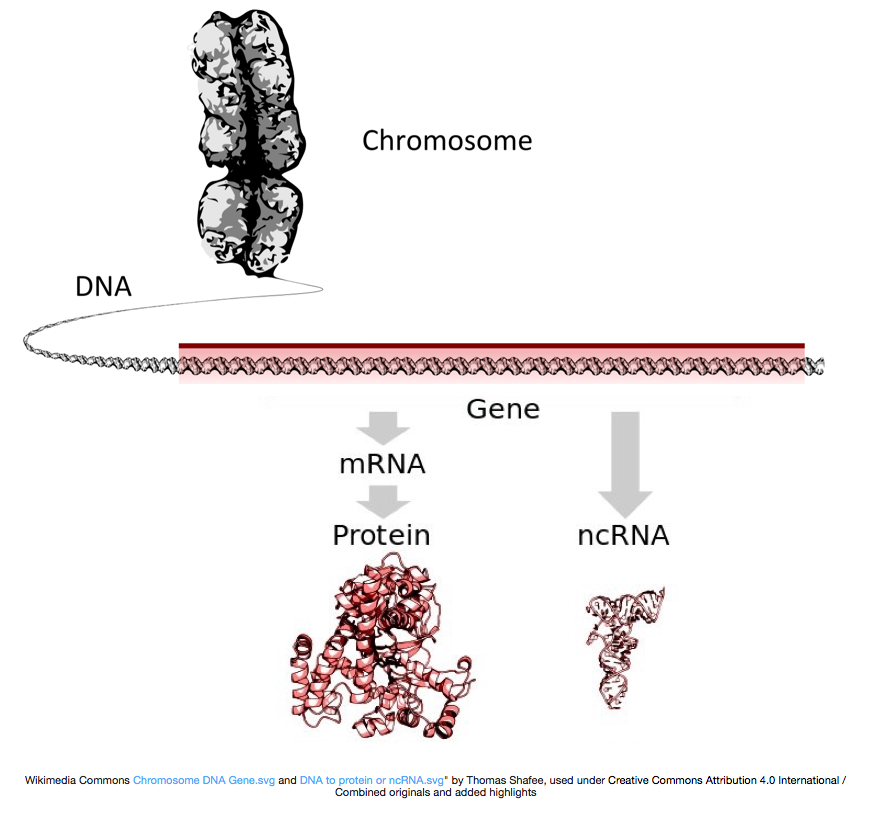
ahhoz, hogy fehérjékké alakuljanak, az RNS-t feldolgozásnak kell alávetni az mRNS előállításához., Az alábbi ábrán a kép felső szála a DNS-ben található gént ábrázolja, amely a le nem fordított régiókból (UTR-ekből) és a nyitott olvasási keretből áll. A géneket előre mRNS-be írják át, amely még mindig tartalmazza az intronikus szekvenciákat. A transzkipciós feldolgozás után az intronok kioldódnak, majd egy Polia farokot és 5 ‘ sapkát adnak hozzá, hogy érett mRNS-átiratokat kapjanak, amelyeket fehérjékké lehet fordítani.,
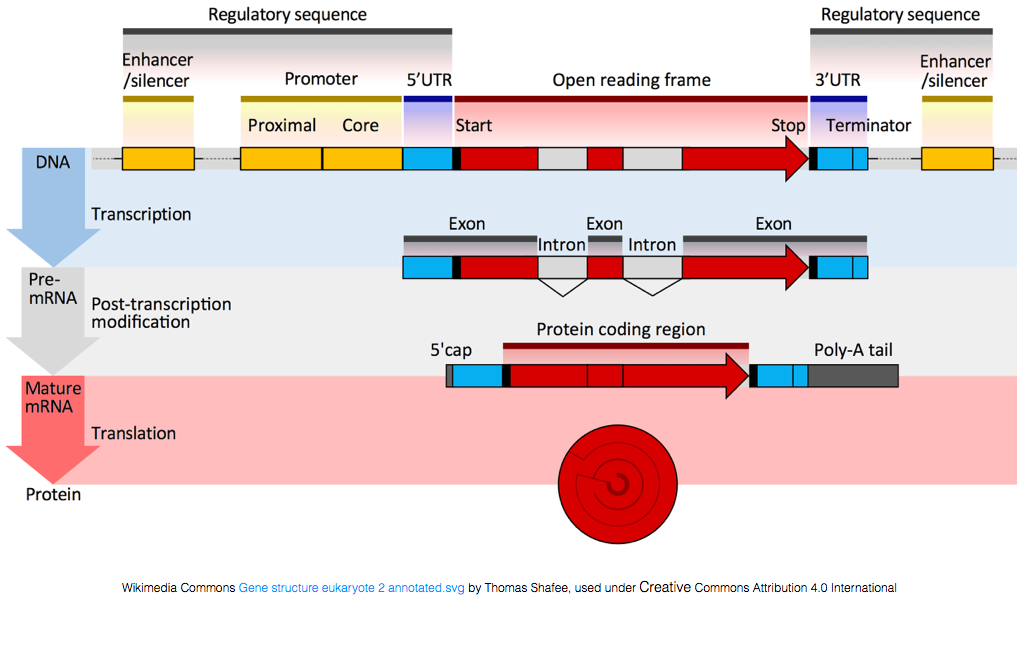
míg az mRNS-átiratok polja farokkal rendelkeznek, sok nem kódoló RNS-átirat nem, mivel a post-transcriptional feldolgozás különbözik ezektől a transzkripcióktól.
Transcriptomics
a transzkriptóma a cellában található összes átirat-leolvasás gyűjteményeként definiálható., Az RNS-seq adatok felhasználhatók egy szervezet transzkriptómájának feltárására és/vagy számszerűsítésére, amelyet a következő típusú kísérletekhez lehet felhasználni:
- differenciál gén expresszió: kvantitatív értékelés és a transzkriptóm szintek összehasonlítása
- transzkriptóm szerelvény: a genom átírt régióinak profiljának felépítése, minőségi értékelés.,
- Lehet használni, hogy segítsen jobban gén modellek ellenőrzik őket, használja a közgyűlés
- Metatranscriptomics vagy közösségi transcriptome elemzés
Illumina könyvtár készítmény
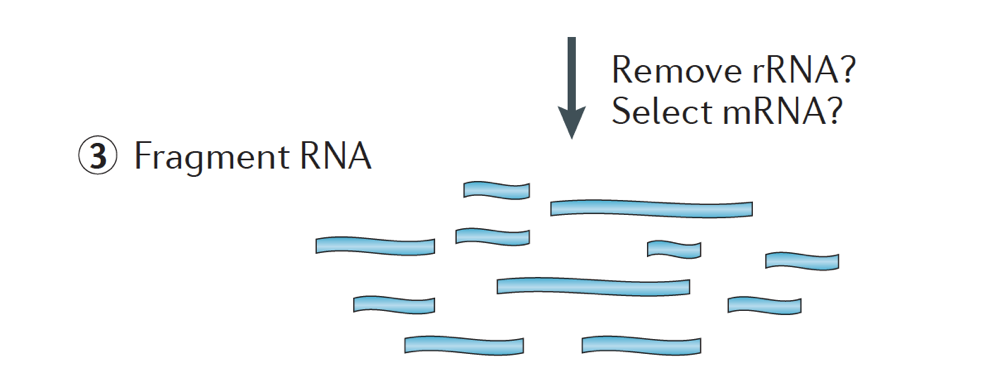
Amikor kezdő RNS-seq kísérlet, minden minta esetében az RNS kell különíteni, majd befordult egy cdns könyvtár sorozatot. A könyvtárkészítés általános munkafolyamatát az alábbi lépésenkénti képek részletezik.
röviden, az RNS izolálódik a mintából, és a DNS-t Dnázzal távolítják el.,
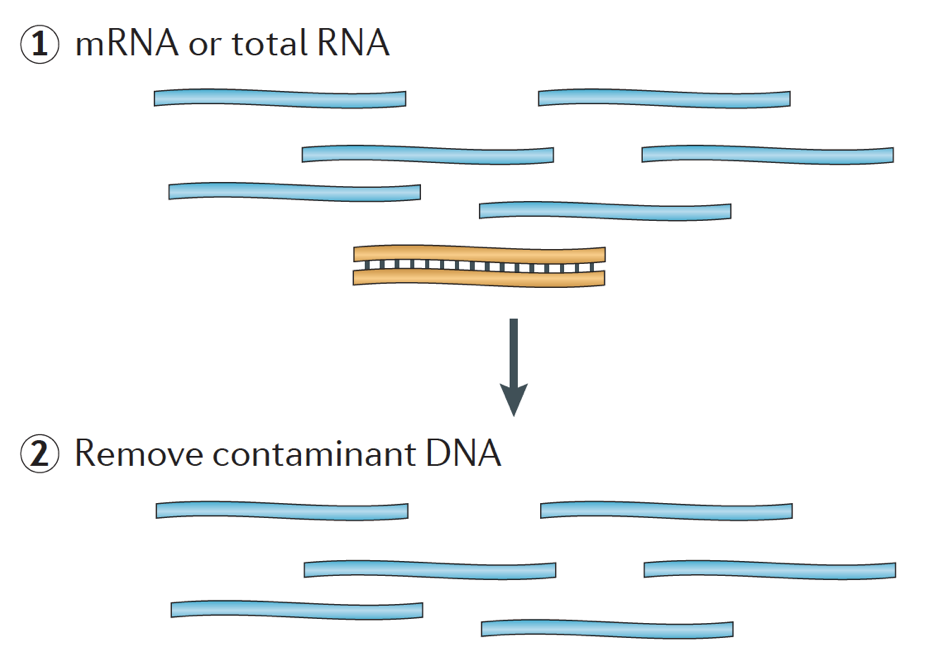
az RNS-minta ezután az mRNS (polyA kiválasztás) vagy az rRNS kimerülésén megy keresztül. A kapott RNS töredezett.
általában a riboszomális RNS képviseli a sejtben jelen lévő RNS-ek többségét, míg a messenger RNS-ek a teljes RNS kis százalékát, ~2% – ot képviselnek emberben. Ezért, ha meg akarjuk vizsgálni a fehérje-kódoló géneket, gazdagítanunk kell az mRNS-t, vagy le kell csökkentenünk az rRNS-t., A differenciál génexpresszió elemzés, a legjobb, hogy gazdagítja a Poly(A)+, kivéve, ha a cél, hogy információt szerezzenek arról, hogy sokáig nem-kódoló Rns, akkor egy ribosomal RNS kimerülése.
a céltöredékek mérete a végső könyvtárban kulcsfontosságú paraméter a könyvtárépítéshez. A DNS-fragmentáció jellemzően fizikai módszerekkel (azaz akusztikus nyírással és szonikációval) vagy enzimatikus módszerekkel (azaz nem specifikus endonukleáz-koktélokkal és transzpozáz tagmentációs reakciókkal) történik.,
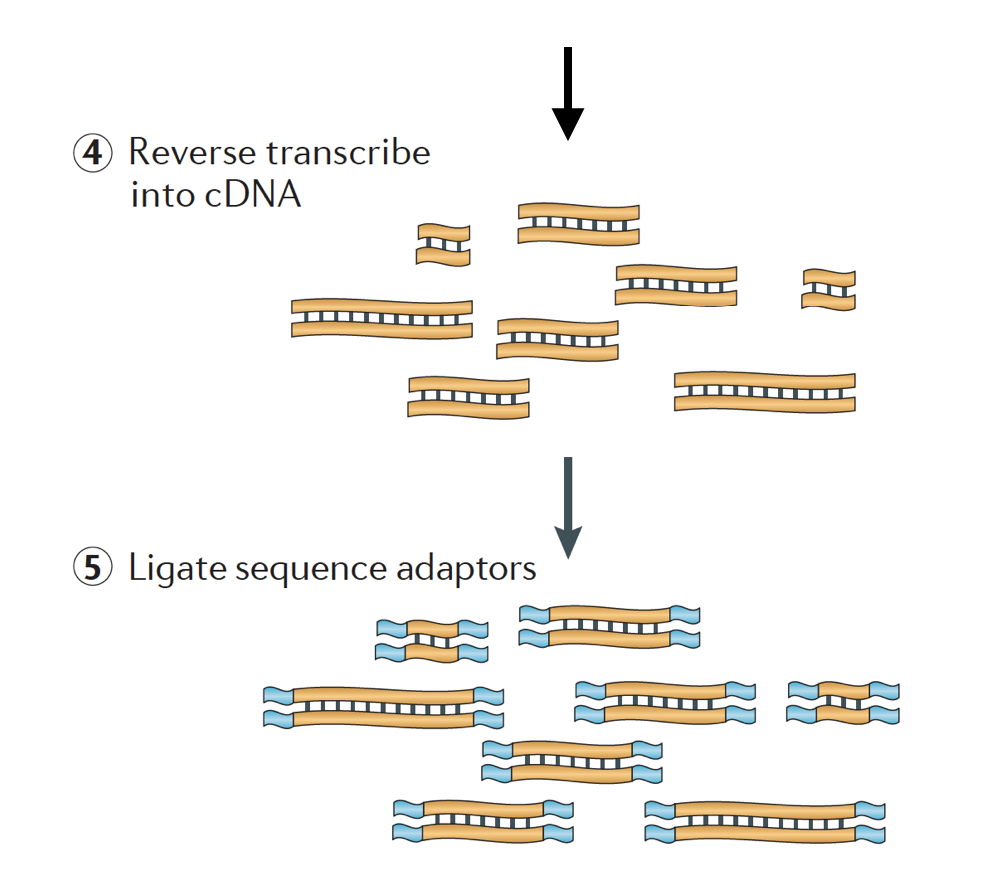
az RNS-t ezután kettős szálú cDNA-ra fordítják, majd a szekvencia adaptereket hozzáadják a töredékek végéhez.
a cDNA könyvtárak olyan módon állíthatók elő, hogy megőrizzék az információt arról, hogy az RNS melyik DNS-szálból származik. Azok a könyvtárak, amelyek megtartják ezt az információt, rekedt könyvtáraknak nevezik, amelyek ma már szabványosak az Illumina TruSeq szálú RNS-Seq készleteivel., A rekedt könyvtárak nem lehetnek drágábbak, mint a nem korlátozottak, ezért nincs igazán ok arra, hogy ne szerezzék meg ezt a kiegészítő információt.,
Van 3 fajta cdns könyvtárak állnak rendelkezésre:
- Előre (secondstrand) – olvas hasonlítanak a gén szekvenciájának vagy a secondstrand cdns szekvencia
- Fordított (firststrand) – olvas hasonlítanak a kiegészítik a gén szekvenciájának vagy firststrand cdns szekvencia (TruSeq)
- Unstranded
Végre, a fragmentumokat PCR erősített, ha szükséges, a fragmentumokat méret kiválasztott (általában ~300-500bp), hogy befejezze a könyvtár.
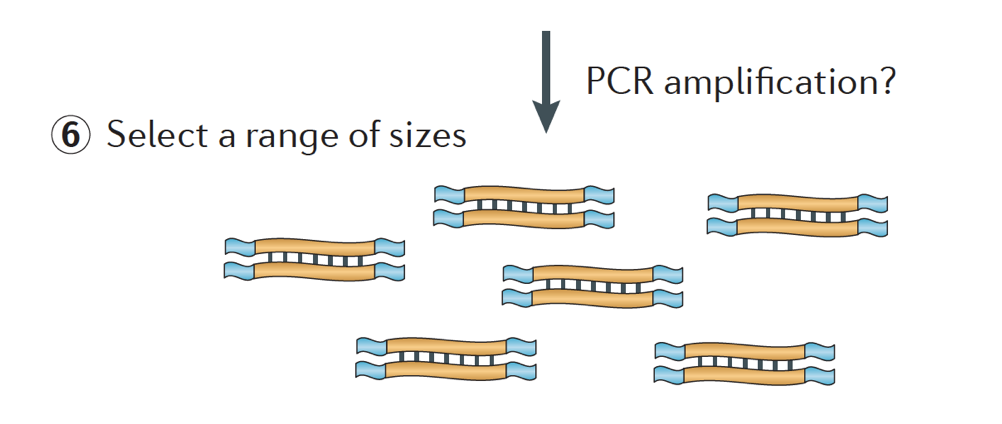
kép hitel: Martin J. A. és Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina szekvenálás
egyvégű versus párosított végű
a könyvtárak elkészítése után szekvenálás végezhető a töredékek végeinek nukleotidszekvenciáinak generálására, amelyeket olvasásnak neveznek. Választhat a cDNA töredékek egyetlen végének szekvenálása (egyvégű olvasás) vagy a töredékek mindkét vége (párosított végolvasás)., 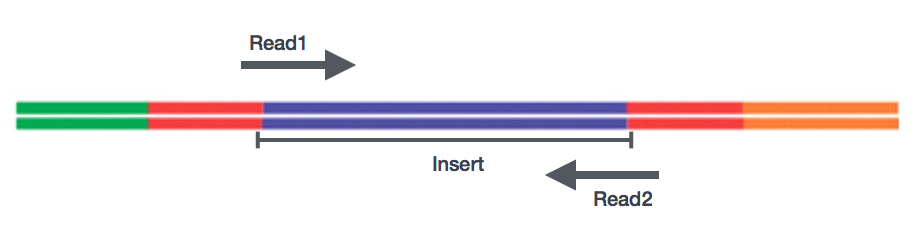
- SE – Egyetlen végén adatkészlet => Csak Read1
- PE – Párosított-end adatkészlet => Read1 + Read2
- lehet 2 külön FastQ fájlokat, vagy csak egy interleaved pár
Általában egy-vége sorozatot, hogy elegendő, ha várható, hogy az olvas lesz meccs, több helyen a genom (pl. szervezetek sok paralogous gének), szerelvények végeznek, vagy a splice izoforma differenciálás. Ne feledje, hogy a párosított végolvasások általában 2x drágábbak.,
különböző szekvenálási platformok
számos Illumina platform közül lehet választani a cDNA könyvtárak sorrendjéhez.

Image credit: adaptált Illumina
különbségek platform megváltoztathatja a hossza olvasás generált, a minőség az olvasás, valamint a teljes számú olvasás szekvenált per futás és a szükséges időt sorrendben a könyvtárak., A különböző platformok mindegyike más áramlási cellát használ, amely egy üvegfelület, amely párosított oligók elrendezésével van bevonva, amelyek kiegészítik a sablonmolekulákhoz hozzáadott adaptereket. Az áramlási cella az, ahol a szekvenálási reakciók zajlanak.

Image credit: Adapted from Illumina
szekvenálás-szintézis
Illumina szekvenálási technológia szekvenálás-szintézis megközelítést alkalmaz, amelyet az alábbiakban részletesebben ismertetünk.
a lépésben a cDNA könyvtár DNS-fragmenseit denaturálják és az üvegáram cellára alkalmazzák., Ezek a denaturált fragmensek kötődnek a komplementer oligókhoz, amelyek már kovalensen kötődnek az áramlási cellasávokhoz, ami kötődést eredményez.
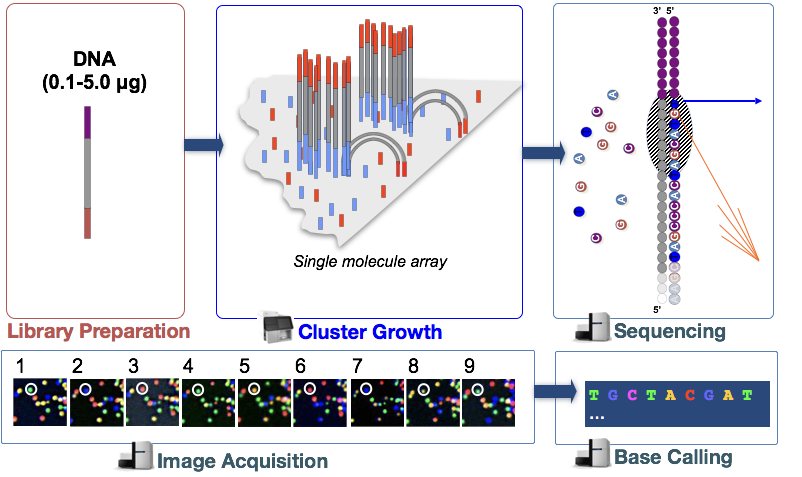
Klasztergeneráció
a töredékek csatolása után megkezdődik a klasztergeneráció nevű fázis. Ebben a lépésben az egyes fragmentumokat klónosan felerősítik, hogy azonos fragmensek klaszterét (fragmenseket) hozzanak létre. Erre azért van szükség, hogy a fluoreszcencia könnyen rögzíthető legyen az egyes klaszterekből, egyetlen fragmens helyett, a nukleotid beépítése során a következő lépésben.,
- Szintetizálni a kiegészítik a polimeráz
- dsDNA denaturált, valamint eredeti DNS-elmosta a víz távozik szintetizált strand covalently köteles áramlási cella.
- egyszálú hibridizálódik a szomszédos adapterrel, így ” híd ”
- a dsDNA polimerázzal bővül. Mindegyik szál kovalensen kötődik a különböző adapterhez.
- ismételje meg többször, hogy klónosan felerősítse az összes egyedi fragmentumot az áramlási cellán, hogy azonos szekvenciájú klasztereket képezzen.,
szekvenálás szintézis útján (& képszerzés)
klaszter generálás után a fluoreszcens címkével ellátott nukleotidokat egyenként (ciklikusan) és fluoreszcens képekkel rögzítik annak meghatározására, hogy melyik nukleotid kerül beépítésre minden egyes ciklusba.
- Denaturációs klaszterek és a 3.blokk ” vége a nem kívánt alapozás megelőzése érdekében.
- Hibridizálja a szekvenáló primereket az illesztő szekvenciához a laza végeken.,
- ciklus négy NTPs fluoreszkáló markerekkel és Terminátor szekvenciával és polimerázokkal.
- az NTP beépítése után a klasztert egy fényforrás gerjeszti, és jellegzetes fluoreszkáló jelet bocsát ki.
- a szín kerül rögzítésre, majd a Terminátor A festék hasítjuk, majd mossuk. A folyamat megismétli a ciklusok meghatározott számát.,
Base Calling
az Illumina saját szoftverrel rendelkezik, amely átmegy az előző szakaszban rögzített összes képen, és szöveges fájlokat generál a fluoreszcencia alapján az egyes klaszterekre vonatkozó sorozatinformációkkal. Amellett, hogy hívja a bázisok, ez a szoftver rendel valószínűségi pontszám jelzi, hogy bizonyos volt a hívás valami “A”, A “T”, A “G”vagy a “C”.
ha vannak kétértelműségek, pl., egy adott ciklusban a klaszter képének nincs olyan különálló színe, amely egy adott nukleotidhoz társulhat, az alaphívó szoftvernek alacsony a valószínűsége, és “A”, “T”, “G” vagy “C” helyett “N” – t rendelne hozzá.
A záró,
- Száma klaszterek ~= Számát olvassa
- Száma szekvenálás ciklus = Hossza olvassa
A ciklusok száma (hossza olvas) függ szekvenálás platform használt, valamint a kedvezményeket.
megjegyzés., Ha mélyebben szeretné felfedezni a szekvenálást szintézis útján, javasoljuk ezt az igazán szép animációt, amely elérhető az Illumina YouTube csatornáján.
Multiplexing
attól függően, hogy az Illumina platform (MiSeq, HiSeq, NextSeq), a sávok száma per flow cella, valamint a számos olvasás, hogy lehet beszerezni egy sáv széles körben változik. El kell döntenie, hogy hány olvasást szeretne mintánként (azaz a szekvenálási mélység), majd a választott platform alapján kiszámítja, hogy hány teljes sávra lesz szüksége a mintakészlethez., Beszéljünk inkább arról, hogy a szempontok, amikor a döntést a következő lecke a Kísérleti Szempontok
Jellemzően díjak a sorozatot, vagy per lane az áramlási cella lesz képes futtatni több minta / lane. Illumina ezért kidolgozott egy szép multiplex módszer, amely lehetővé teszi a könyvtárak több mintát kell összevonni és szekvenált egyszerre ugyanabban a sávban egy áramlási cella. Ez a módszer indexek (az Illumina adapteren belül) vagy speciális vonalkódok (az Illumina adapteren kívül) hozzáadását igényli az alábbi vázlatban leírtak szerint.,
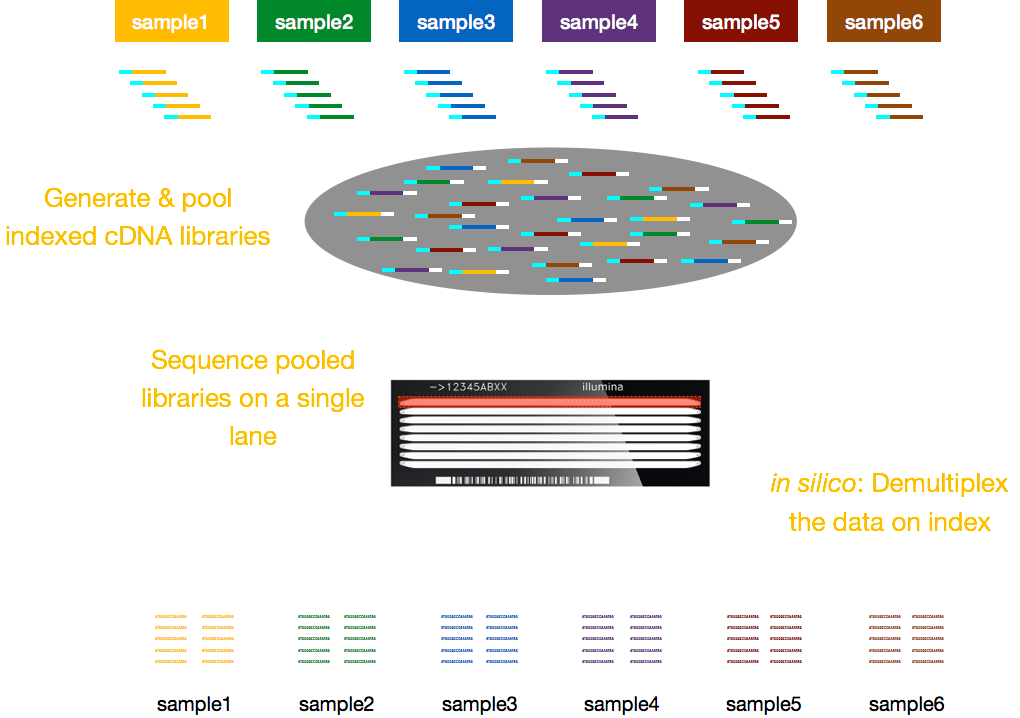
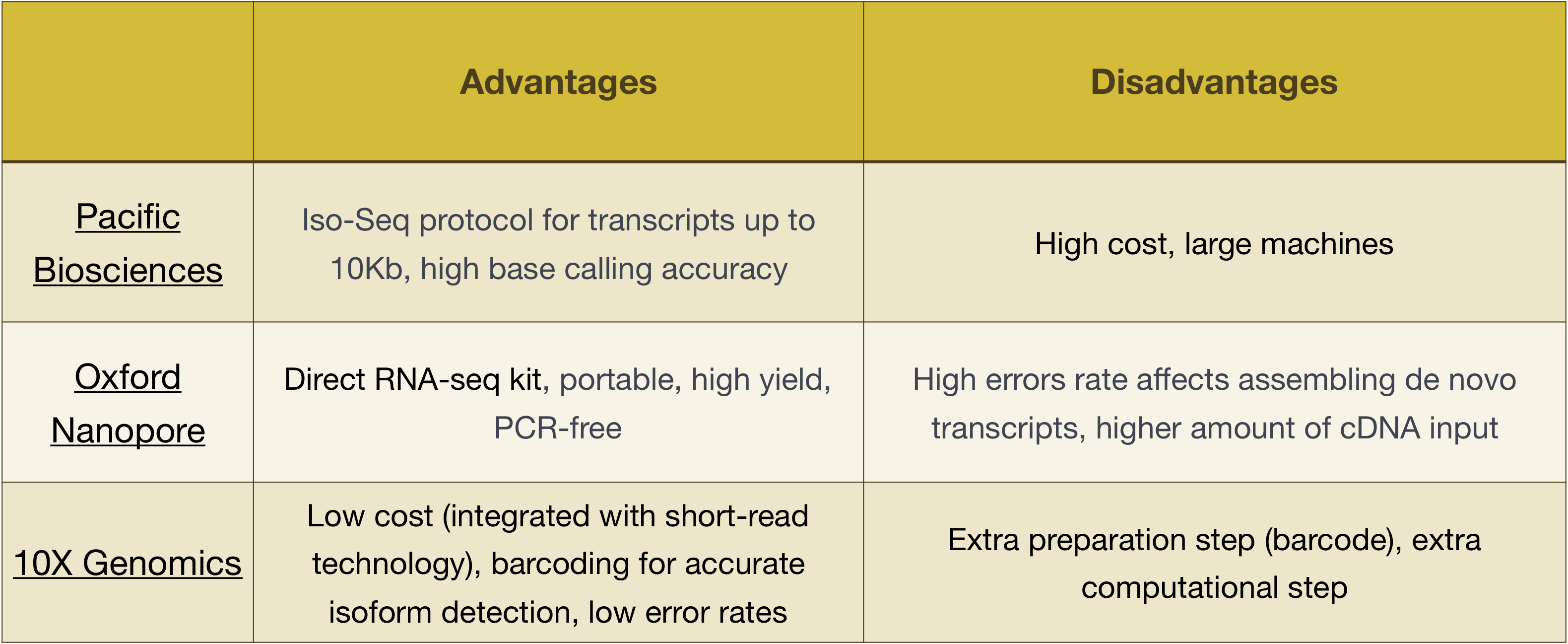
Megjegyzés: Az ebben a leckében bemutatott munkafolyamat az Illumina szekvenálásra jellemző, amely jelenleg a leginkább használt szekvenálási módszer., De vannak egyéb hosszú-olvasd el a sorozatot, hogy a módszereket érdemes megjegyezni, például:
- Csendes-óceáni Biosciences: http://www.pacb.com/
- Oxford Nanopore (Szolga): https://nanoporetech.com/
- 10X Genomika: https://www.10xgenomics.com/
Előnyök, hátrányok ezek a technológiák is meg kell vizsgálni az alábbi táblázatban:
Vélemény, hozzászólás?