Approssimativa: 90 minuti
Obiettivi di Apprendimento:
- Descrivere il processo di RNA-seq la preparazione di libreria
- Descrivere il metodo di sequenziamento Illumina
Introduzione di RNA-seq
RNA-seq è un eccitante tecnica sperimentale che viene utilizzato per esplorare e/o quantificare l’espressione genica all’interno o tra le condizioni.,
Come sappiamo, i geni forniscono istruzioni per produrre proteine, che svolgono alcune funzioni all’interno della cellula. Sebbene tutte le cellule contengano la stessa sequenza di DNA, le cellule muscolari sono diverse dalle cellule nervose e da altri tipi di cellule a causa dei diversi geni attivati in queste cellule e dei diversi RNA e proteine prodotti.
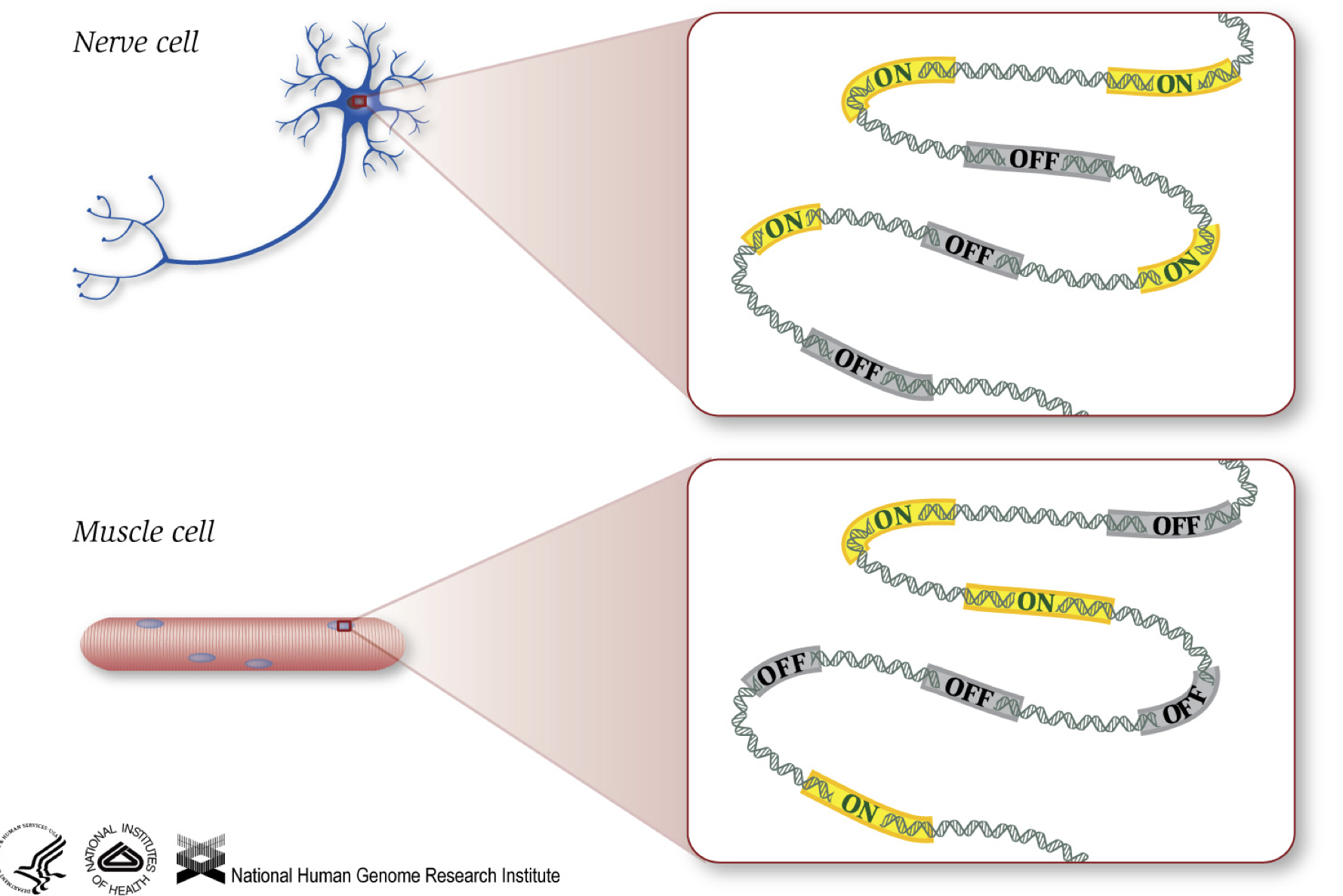
Diversi processi biologici, così come le mutazioni, possono influenzare quali geni sono accesi e quali sono spenti, oltre a, quanti geni specifici sono accesi / spenti.,
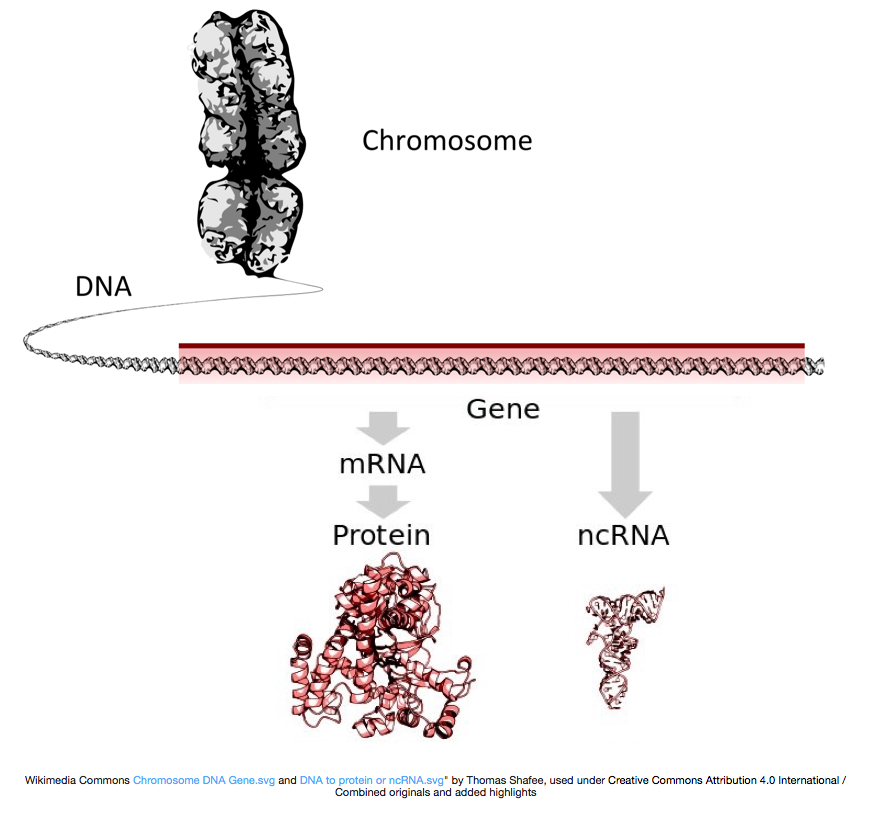
Per produrre proteine, il DNA viene trascritto in RNA messaggero, o mRNA, che viene tradotto dal ribosoma in proteina. Tuttavia, alcuni geni codificano l’RNA che non viene tradotto in proteine; questi RNA sono chiamati RNA non codificanti o NCRNA. Spesso questi RNA hanno una funzione in sé e per sé e includono RRNA, TRNA e SIRNA, tra gli altri. Tutti gli RNA trascritti dai geni sono chiamati trascritti.
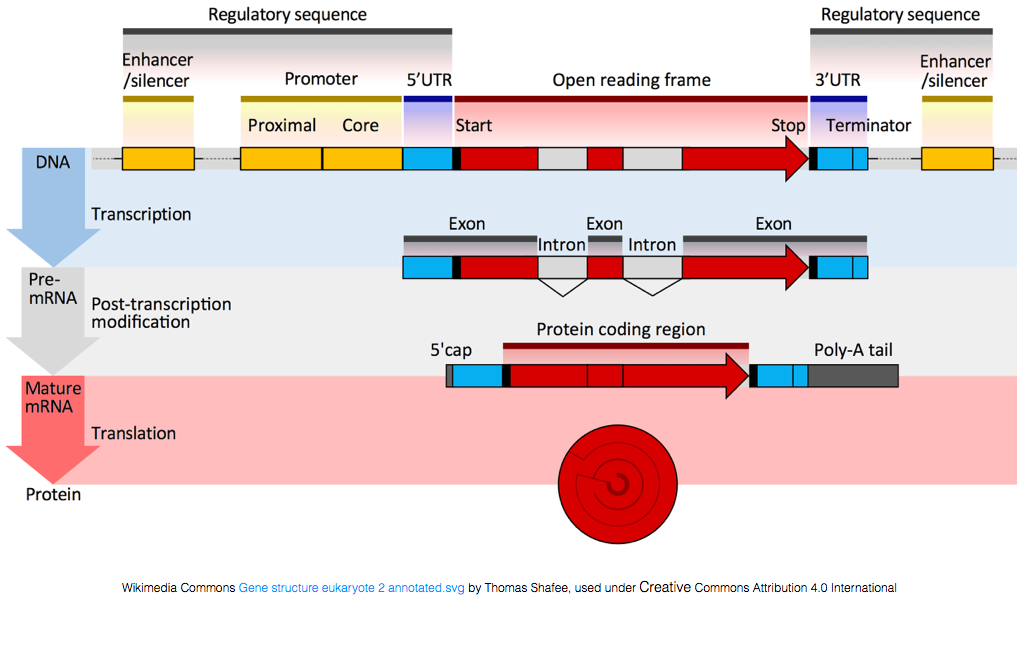
Per essere tradotto in proteine, l’RNA deve essere sottoposto ad elaborazione per generare l’mRNA., Nella figura seguente, il filamento superiore nell’immagine rappresenta un gene nel DNA, composto dalle regioni non tradotte (UTRs) e dal frame di lettura aperto. I geni sono trascritti in pre-mRNA, che contiene ancora le sequenze introniche. Dopo l’elaborazione post-transciptional, gli introni sono impiombati fuori e una coda di polyA ed un cappuccio 5 ‘ sono aggiunti per produrre i trascritti maturi del mRNA, che possono essere tradotti in proteine.,
Mentre le trascrizioni di mRNA hanno una coda polyA, molte delle trascrizioni di RNA non codificanti non lo fanno poiché l’elaborazione post-trascrizionale è diversa per queste trascrizioni.
Transcriptomics
Il transcriptome è definito come una raccolta di tutte le letture di trascrizione presenti in una cella., Dati di RNA-seq può essere utilizzato per esplorare e/o quantificare il trascrittoma di un organismo, che può essere utilizzato per i seguenti tipi di esperimenti:
- Espressione Genica Differenziale: valutazione quantitativa e il confronto dei livelli di trascrizione
- Transcriptome montaggio: costruzione del profilo del trascritto regioni del genoma, una valutazione qualitativa.,
- Può essere usato per aiutare a costruire modelli genici migliori e verificarli usando l’assembly
- Metatranscriptomics o community transcriptome analysis
Illumina library preparation
Quando si avvia un esperimento RNA-seq, per ogni campione l’RNA deve essere isolato e trasformato in una libreria cDNA per il sequenziamento. Il flusso di lavoro generale per la preparazione della libreria è dettagliato nelle immagini passo-passo di seguito.
In breve, l’RNA viene isolato dal campione e il DNA contaminante viene rimosso con DNasi.,
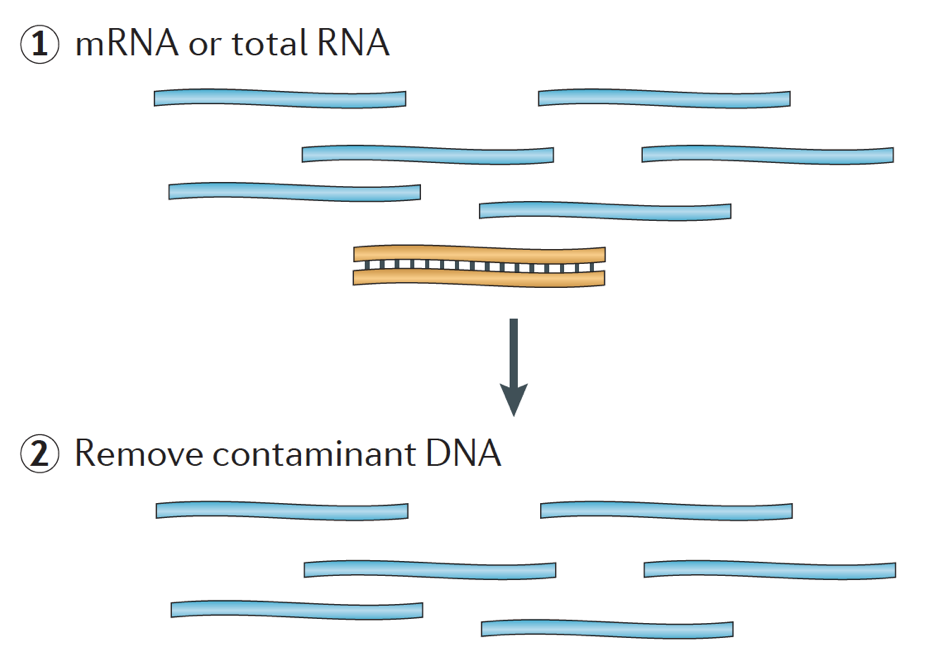
Il campione di RNA subisce quindi la selezione dell’mRNA (selezione polyA) o l’esaurimento dell’rRNA. L’RNA risultante è frammentato.
Generalmente, l’RNA ribosomiale rappresenta la maggior parte degli RNA presenti in una cellula, mentre gli RNA messaggeri rappresentano una piccola percentuale di RNA totale, ~2% nell’uomo. Pertanto, se vogliamo studiare i geni che codificano le proteine, dobbiamo arricchire l’mRNA o esaurire l’rRNA., Per l’analisi dell’espressione genica differenziale, è meglio arricchire per Poly (A)+, a meno che non si miri a ottenere informazioni sugli RNA non codificanti lunghi, quindi eseguire una deplezione di RNA ribosomiale.
La dimensione dei frammenti di destinazione nella libreria finale è un parametro chiave per la costruzione della libreria. La frammentazione del DNA viene tipicamente eseguita con metodi fisici (es. taglio acustico e sonicazione) o metodi enzimatici (es. cocktail endonucleasici non specifici e reazioni di tagmentazione della trasposasi.,
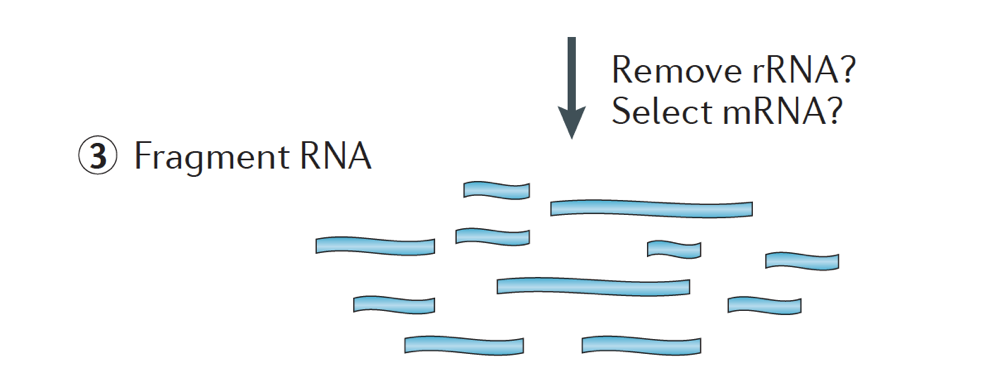
L’RNA viene quindi trascritto in cDNA a doppio filamento e gli adattatori di sequenza vengono quindi aggiunti alle estremità dei frammenti.
Le librerie cDNA possono essere generate in modo da conservare le informazioni su quale filamento di DNA è stato trascritto l’RNA. Le librerie che conservano queste informazioni sono chiamate librerie stranded, che ora sono standard con i kit Truseq stranded RNA-Seq di Illumina., Le librerie Stranded non dovrebbero essere più costose di unstranded, quindi non c’è davvero alcun motivo per non acquisire queste informazioni aggiuntive.,
Ci sono 3 tipi di librerie di cDNA disponibili:
- Avanti (secondstrand) – legge simile sequenza genica o la secondstrand sequenza di cDNA
- Inverti (firststrand) – legge simile il completamento della sequenza del gene o firststrand sequenza di cDNA (TruSeq)
- Unstranded
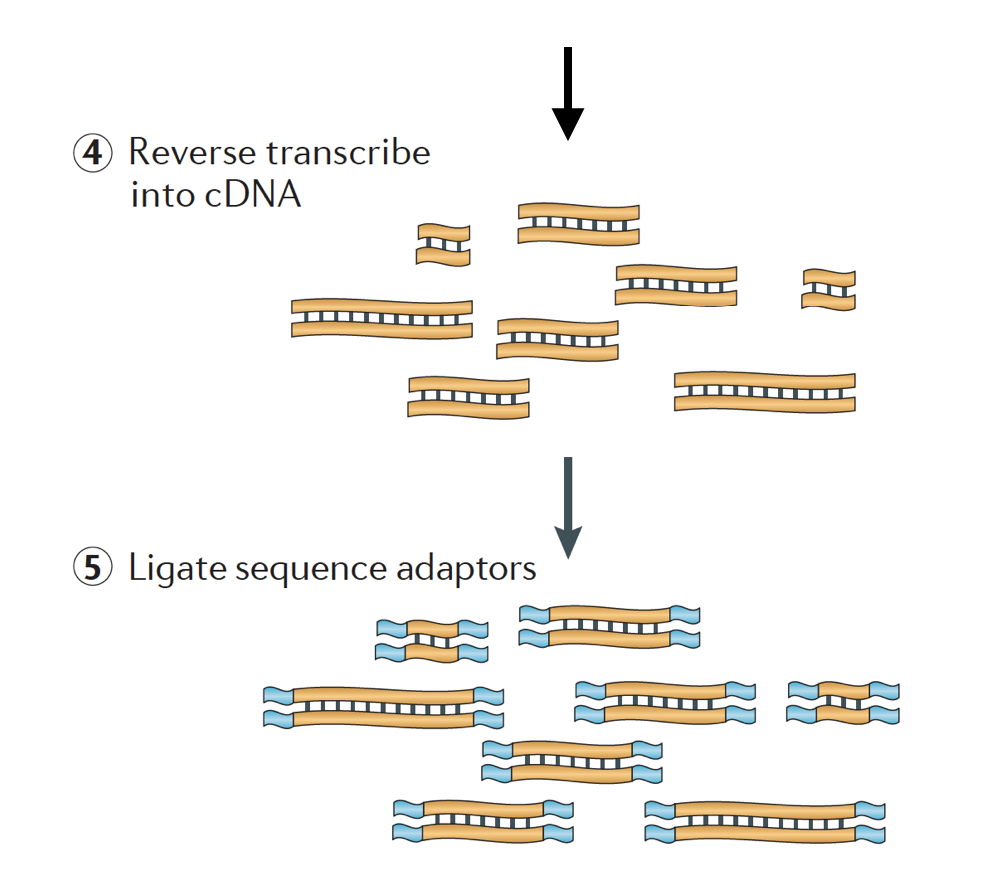
Infine, i frammenti di PCR amplificato se necessario, e i frammenti delle dimensioni di (di solito ~300-500bp) per completare la raccolta.
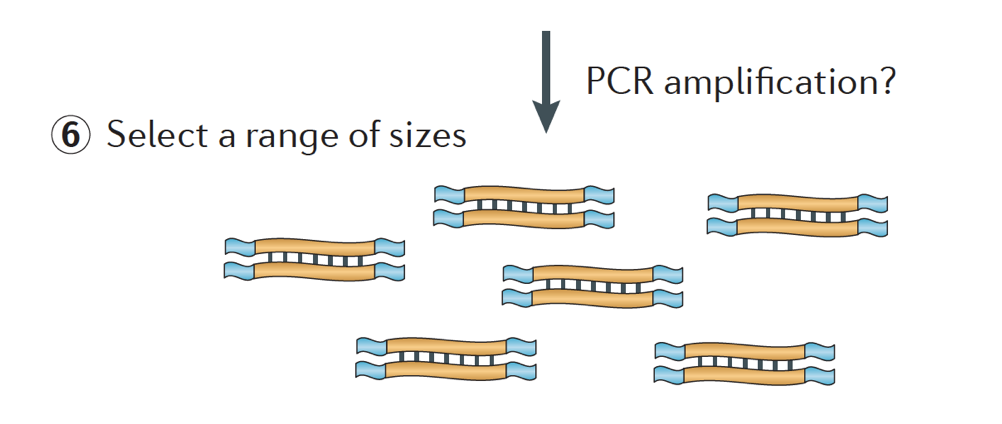
Immagine di credito: Martin J. A. e Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina Sequencing
Single-end versus Paired-end
Dopo la preparazione delle librerie, il sequenziamento può essere eseguito per generare le sequenze nucleotidiche delle estremità dei frammenti, che sono chiamate letture. Avrete la possibilità di sequenziare una singola estremità dei frammenti cDNA (single-end legge) o entrambe le estremità dei frammenti (accoppiati-end legge).,
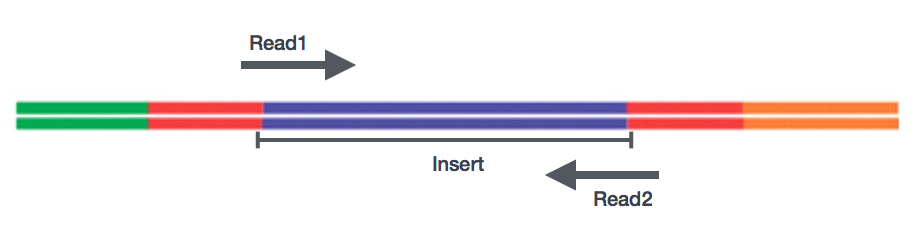
- SE – Unico fine dataset => Solo Read1
- PE – Paired-end dataset => Read1 + Read2
- può essere separate per 2 FastQ file o solo con interleaved coppie
in genere unico fine di sequenziamento è sufficiente se non è previsto che la legge corrispondono di più sedi sul genoma (ad esempio organismi con molti geni paralogous), le assemblee sono state effettuate, o per incollaggio isoforma di differenziazione. Tieni presente che le letture accoppiate sono generalmente 2 volte più costose.,
Diverse piattaforme di sequenziamento
Ci sono una varietà di piattaforme Illumina tra cui scegliere per sequenziare le librerie cDNA.

Credito immagine: Adattato da Illumina
Le differenze nella piattaforma possono alterare la lunghezza delle letture generate, la qualità delle letture, così come il numero totale di letture sequenziate per esecuzione e la quantità di tempo necessario per sequenziare le librerie., Le diverse piattaforme utilizzano ciascuna una cella di flusso diversa, che è una superficie di vetro rivestita con una disposizione di oligos accoppiati che sono complementari agli adattatori aggiunti alle molecole del modello. La cella di flusso è dove avvengono le reazioni di sequenziamento.

Immagine di credito: Adattato da Illumina
Sequencing-by-synthesis
Illumina tecnologia di sequenziamento utilizza un approccio sequencing-by-synthesis che è descritto in modo più dettagliato di seguito.
Nel passaggio, i frammenti di DNA nella libreria cDNA vengono denaturati e applicati alla cella di flusso di vetro., Questi frammenti denaturati si legano agli oligos complementari che sono già legati covalentemente alle corsie delle cellule di flusso, con conseguente attaccamento.
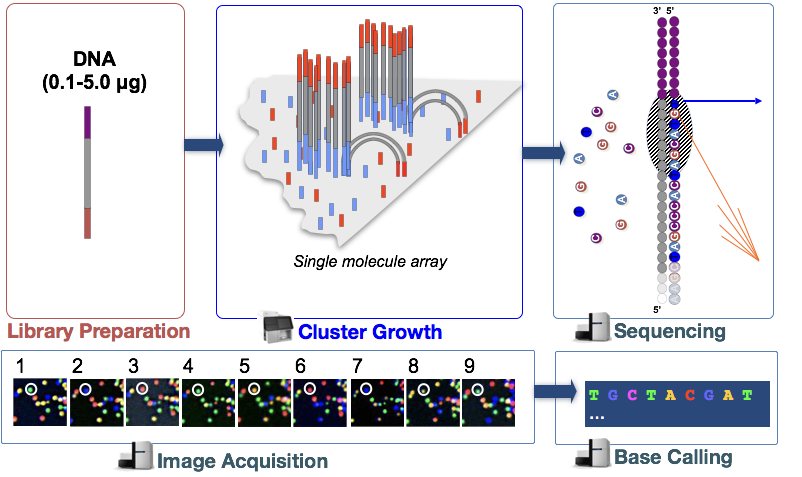
Generazione cluster
Una volta che i frammenti si sono collegati, inizia una fase chiamata generazione cluster. Durante questa fase, singoli frammenti vengono clonalmente amplificati per creare un cluster (frammenti in prossimità) di frammenti identici. Ciò è necessario in modo che la fluorescenza possa essere prontamente catturata da ciascun cluster, invece di un singolo frammento, durante l’incorporazione del nucleotide nella fase successiva.,
- Sintetizzare il complemento con polimerasi
- dsDNA è denaturato, e DNA originale lavato via lasciando sintetizzato filo covalentemente legato alla cella di flusso.
- Il singolo filo si ibrida con l’adattatore adiacente per formare un ‘ponte’
- dsDNA è esteso dalla polimerasi. Ogni filo legato in modo covalente a un adattatore diverso.
- Ripeti molte volte per amplificare clonalmente tutti i frammenti unici sulla cella di flusso per formare cluster di sequenza identica.,
Sequenziamento per sintesi (& acquisizione di immagini)
Dopo cluster generazione, fluorescente contrassegnati nucleotidi sono incorporate una alla volta (ciclicamente) e le immagini di fluorescenza vengono catturati per identificare il nucleotide è incorporato in ogni cluster in ogni ciclo.
- Denatura cluster e il blocco 3’ estremità per evitare adescamento indesiderato.
- Ibridare sequenziamento primer per adattatore sequenza alle estremità libere.,
- Ciclo quattro NTPS con marcatori fluorescenti e sequenza terminatrice e polimerasi.
- Una volta incorporato l’NTP, il cluster viene eccitato da una sorgente luminosa e viene emesso un caratteristico segnale fluroscente.
- Il colore viene registrato, quindi il terminatore sulla tintura viene scisso e lavato. Il processo si ripete per il numero specificato di cicli.,
Base Calling
Illumina ha un software proprietario che passa attraverso tutte le immagini catturate nella fase precedente e genera file di testo con informazioni di sequenza su ogni cluster in base alla fluorescenza. Oltre a chiamare le basi, questo software assegna un punteggio di probabilità per indicare quanto fosse certo di chiamare qualcosa di ” A”, “T”, “G”o “C”.
Se ci sono ambiguità, ad esempio, ad un certo ciclo l’immagine per un cluster non ha un colore distinto che può essere associato a un nucleotide specifico, il software di chiamata di base avrà una bassa probabilità associata ad esso e assegnerebbe una “N” invece di “A”, “T”, “G” o “C”.
chiusura
- Numero di cluster ~= Numero di letture
- Numero di cicli di sequenziamento = Lunghezza della legge
Il numero di cicli (lunghezza della legge) dipenderà dalla piattaforma di sequenziamento utilizzati così come le vostre preferenze.
NOTA., Se vuoi esplorare il sequenziamento per sintesi in modo più approfondito, ti consigliamo questa animazione davvero piacevole disponibile sul canale YouTube di Illumina.
Multiplexing
A seconda della piattaforma Illumina (MiSeq, HiSeq, NextSeq), il numero di corsie per cella di flusso e il numero di letture che possono essere ottenute per corsia varia ampiamente. Dovrai decidere quante letture desideri per campione (cioè la profondità di sequencning) e quindi in base alla piattaforma scelta calcola quante corsie totali richiederai per il tuo set di campioni., Parleremo di più sulle considerazioni quando si prende questa decisione nella prossima lezione sulle considerazioni sperimentali
In genere, le spese per il sequenziamento sono per corsia della cella di flusso e si sarà in grado di eseguire più campioni per corsia. Illumina ha quindi ideato un bel metodo di multiplexing che consente alle librerie di diversi campioni di essere raggruppate e sequenziate contemporaneamente nella stessa corsia di una cella di flusso. Questo metodo richiede l’aggiunta di indici (all’interno dell’adattatore Illumina) o codici a barre speciali (all’esterno dell’adattatore Illumina) come descritto nello schema seguente.,
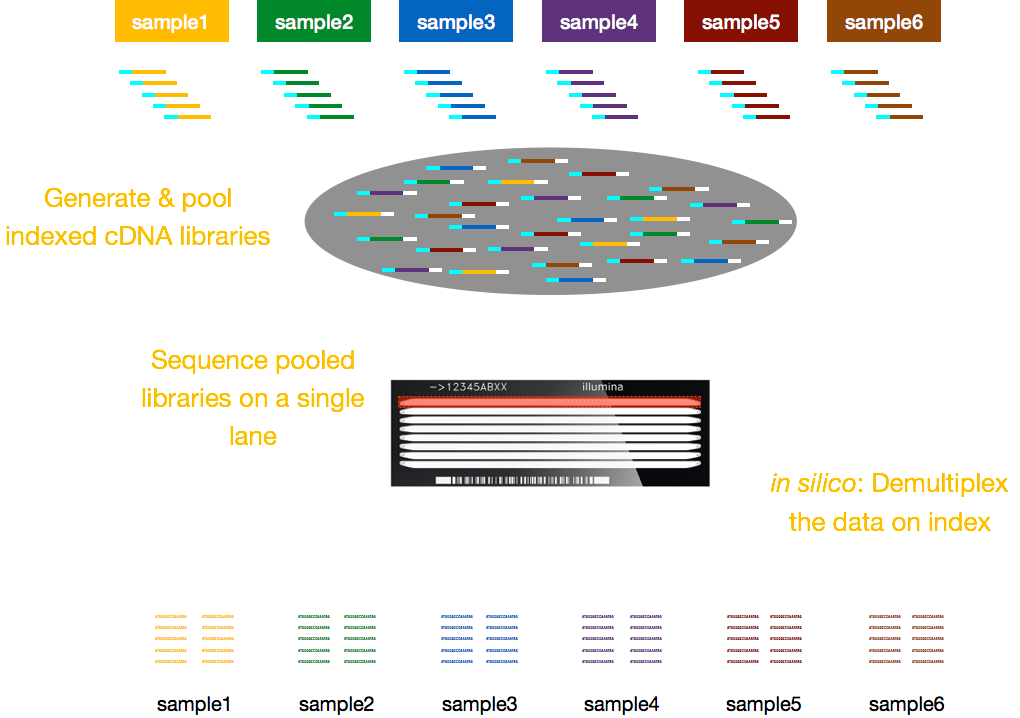
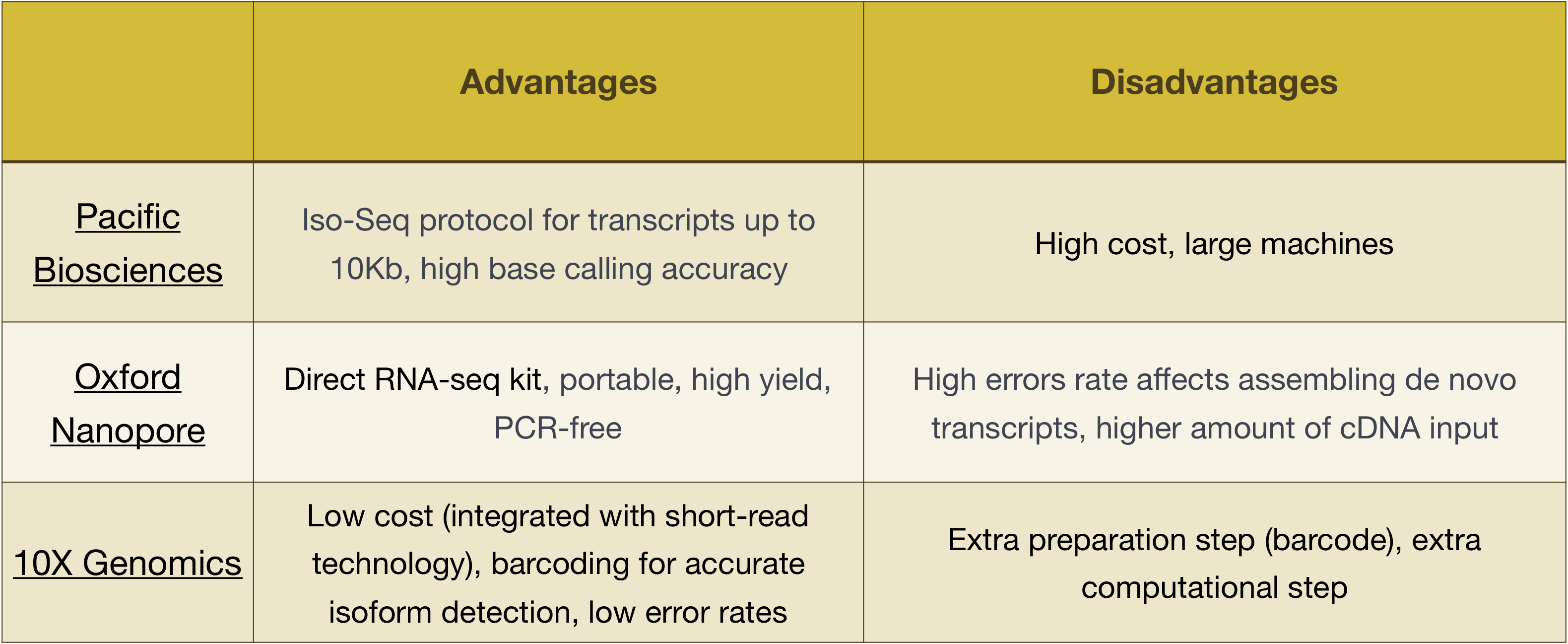
NOTA: Il flusso di lavoro presentato in questa lezione è specifico per Illumina sequencing, che è attualmente il metodo di sequenziamento più utilizzato., Ma ci sono altre leggi di metodi di sequenziamento pena di notare, come:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomica: https://www.10xgenomics.com/
i Vantaggi e gli svantaggi di queste tecnologie può essere esplorato nella tabella di seguito:
Lascia un commento