Omtrentlig tid: 90 minutter
læringsmål:
- Beskrive prosessen av RNA-seq bibliotek forberedelse
- Beskrive Illumina-sekvensering metode
Introduksjon til RNA-seq
RNA-seq er en spennende eksperimentell teknikk som brukes til å utforske og/eller kvantifisere genuttrykk innen eller mellom forhold.,
Som vi vet, gener gir instruksjoner for å lage proteiner, som utfører noen funksjon i cellen. Selv om alle cellene inneholder det samme DNA sekvens, muskel celler er forskjellige fra nerveceller og andre typer celler på grunn av ulike gener som er skrudd på i disse cellene og de ulike RNAs og proteiner som produseres.
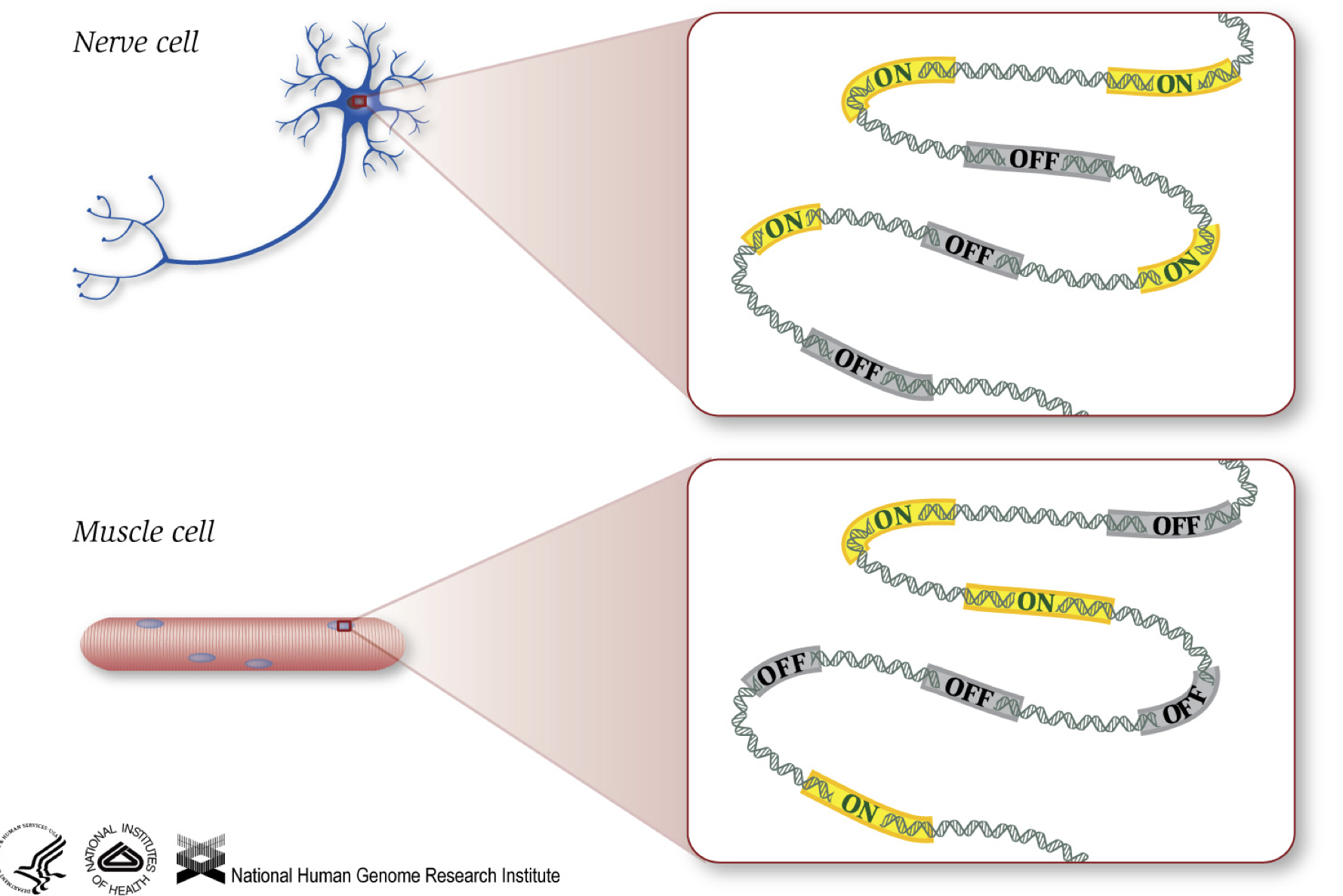
Forskjellige biologiske prosesser, samt mutasjoner, kan påvirke hvilke gener som er slått på, og som er slått av, i tillegg til, hvor mye spesifikke gener er slått på/slått av.,
for Å lage proteiner, DNA kopieres inn i messenger-RNA, eller mRNA, som er oversatt av ribosomet til protein. Imidlertid, noen gener som koder for RNA som ikke får oversatt til proteiner, og disse RNAs er kalt ikke-kodende RNAs, eller ncRNAs. Ofte er disse RNAs har en funksjon i seg selv og inkluderer rRNAs, tRNAs, og siRNAs, blant andre. Alle RNAs transkribert fra genene kalles transkripsjoner.
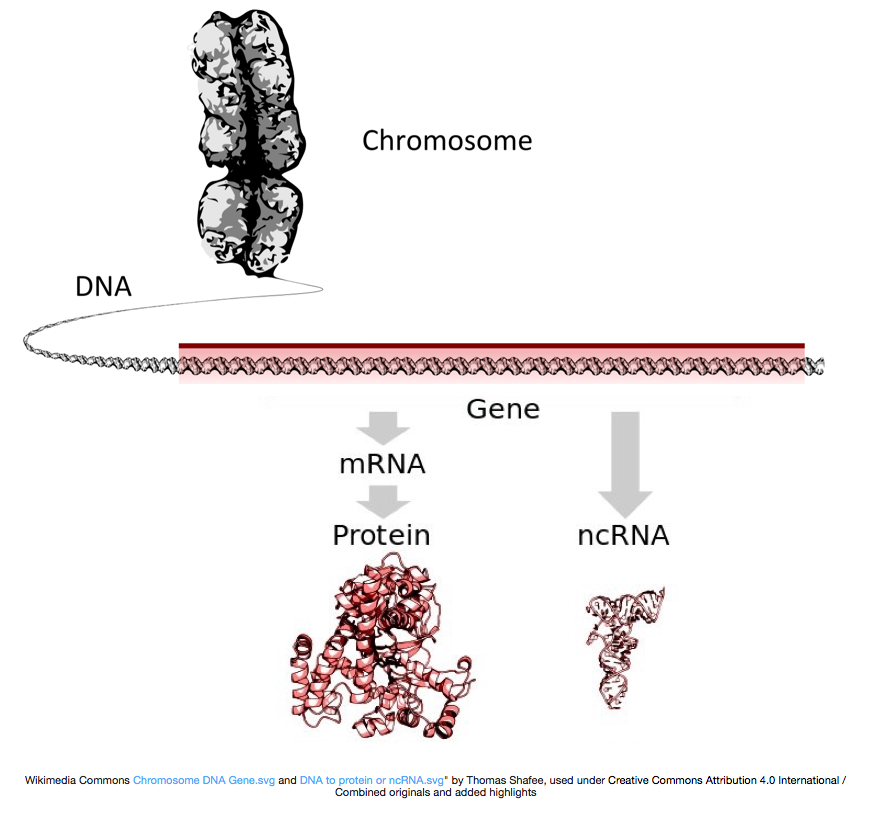
for Å bli oversatt til proteiner, RNA må gjennomgå behandling for å generere mRNA., I figuren nedenfor toppen strand i bildet representerer en genet i DNA, som består av uoversatt regioner (UTRs) og åpne lese ramme. Gener er transkribert i pre-mRNA, som fortsatt inneholder intronic sekvenser. Etter post-transciptional prosessering, introns er splittet ut og en polyA halen og 5′ cap er lagt for å gi modne mRNA transkripter, som kan bli oversatt til proteiner.,
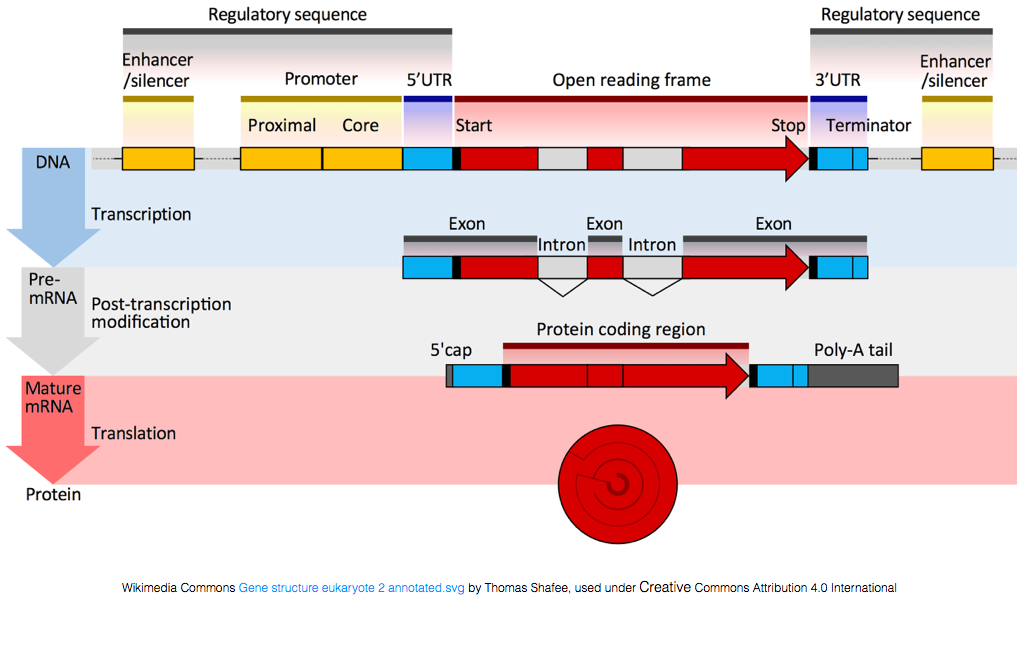
Mens mRNA transkripter har en polyA halen, mange av de ikke-kodende RNA transkripsjoner ikke som post-transcriptional behandling er forskjellig for disse transkripsjonene.
Transcriptomics
transcriptome er definert som en samling av alle transkripsjonen avlesning til stede i en celle., RNA-seq data kan brukes til å utforske og/eller kvantifisere transcriptome av en organisme, som kan benyttes for følgende typer eksperimenter:
- Differensiert genuttrykk: kvantitativ evaluering og sammenligning av transkripsjonen nivåer
- Transcriptome montering: bygge profil av transkribert regioner av genomet, en kvalitativ evaluering.,
- Kan brukes til å bidra til å bygge bedre genet modeller, og kontrollerer dem ved hjelp av forsamlingen
- Metatranscriptomics eller samfunnet transcriptome analyse
Illumina bibliotek forberedelse
Når du starter et RNA-seq-eksperiment, for hver prøve RNA behov for å være isolert og omgjort til et cDNA-bibliotek for sekvensering. Den generelle arbeidsflyt for bibliotek forberedelse er detaljert beskrevet i trinn-for-trinn-bilder nedenfor.
Kort, RNA isolert fra prøven og kontaminerende DNA er fjernet med DNase.,
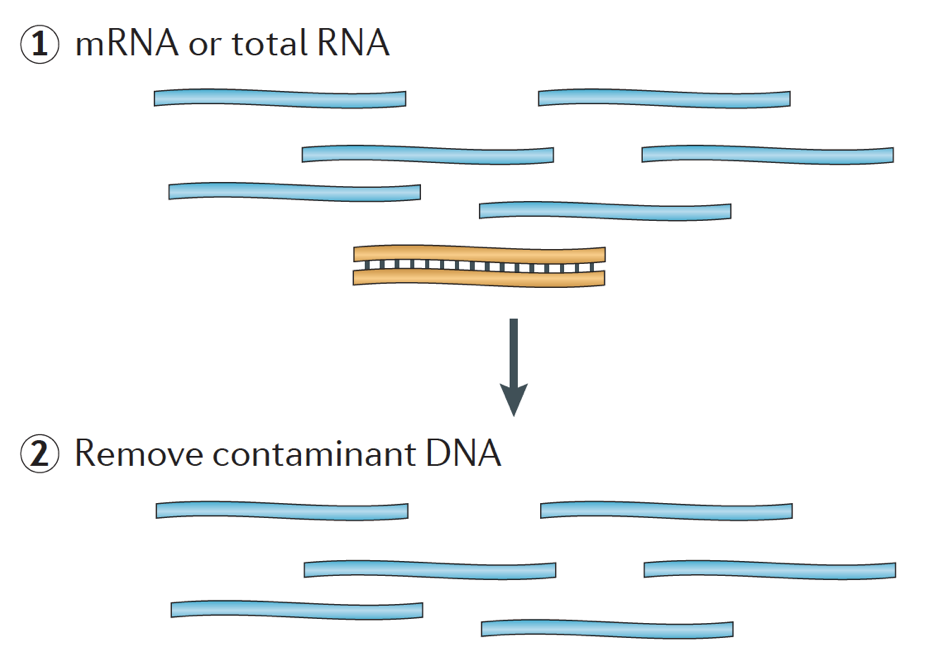
RNA eksempel deretter gjennomgår enten utvalg av mRNA (polyA utvalget) eller uttømming av rRNA. Den resulterende RNA er fragmentert.
Vanligvis, ribosom-RNA representerer flertallet av RNAs til stede i en celle, mens messenger RNAs representerer en liten andel av total RNA, ~2% i mennesker. Derfor, hvis vi ønsker å studere protein-kodende gener, vi trenger å berike for mRNA eller dreper rRNA., For differensiert genuttrykk analyse, er det best å berike for Poly(A)+, med mindre du sikter til å få informasjon om lange ikke-kodende RNAs, og deretter gjøre en ribosom-RNA-forbruk.
størrelsen på målet fragmenter i den endelige biblioteket er en viktig parameter for bibliotek konstruksjon. DNA-fragmentering er vanligvis gjort ved fysiske metoder (dvs., akustisk shearing og sonikering) eller enzymatisk metoder (dvs., ikke-spesifikk endonuclease cocktailer og transposase tagmentation reaksjoner.,
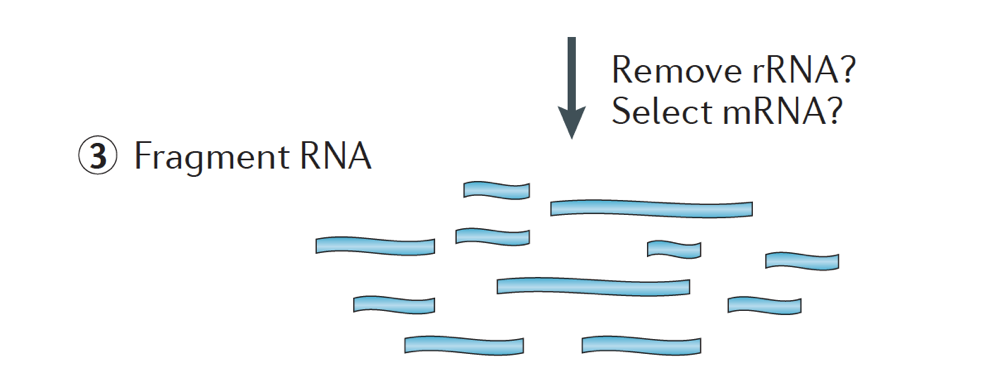
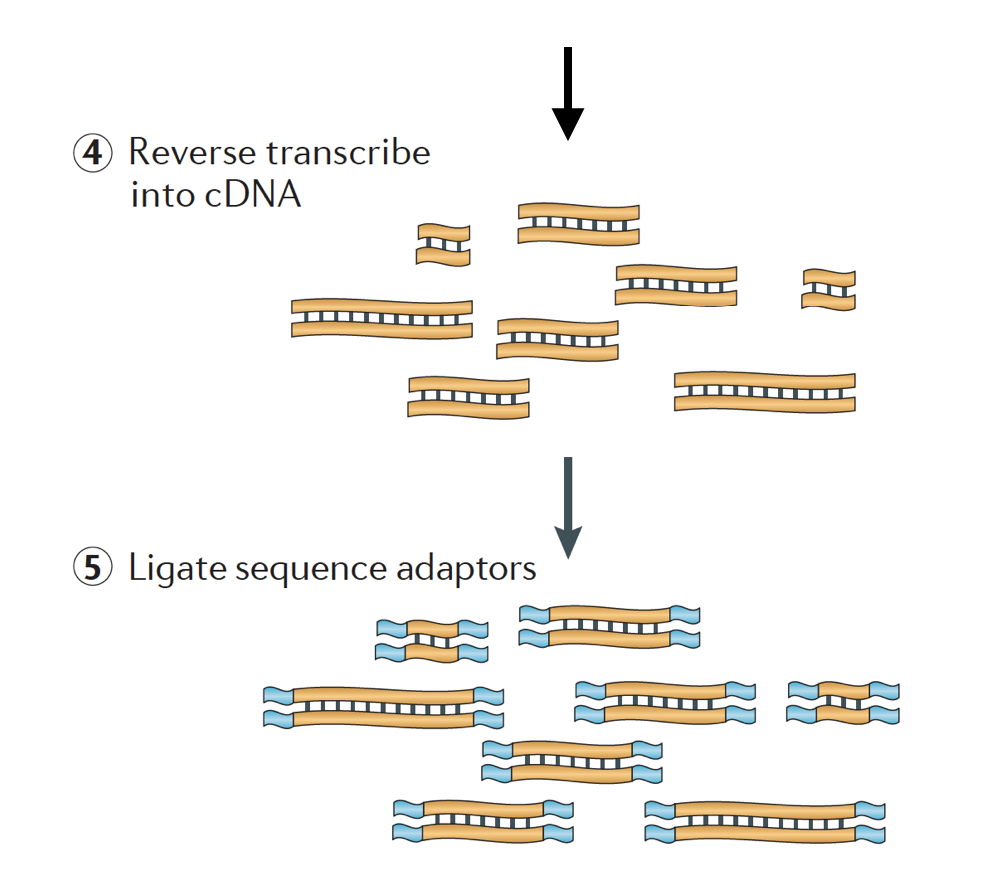
RNA er da omvendt skrevet inn dobbel-strandet cDNA og sekvens-adaptere er da lagt til endene av fragmenter.
cDNA biblioteker kan genereres på en måte å ta vare på informasjon om hvilke strand av DNA, RNA ble transkribert fra. Biblioteker som lagrer denne informasjonen kalles strandet bibliotekene, som nå er standard med Illumina er TruSeq strandet RNA-Seq-kits., Strandet bibliotekene bør ikke være noe dyrere enn unstranded, så det er egentlig ikke noen grunn til ikke å skaffe seg denne informasjonen.,
Det er 3 typer av cDNA biblioteker tilgjengelig:
- Videresend (secondstrand) – leser ligne genet sekvens eller secondstrand cDNA rekkefølge
- Omvendt (firststrand) – leser ligne bemanning av genet sekvens eller firststrand cDNA sekvens (TruSeq)
- Unstranded
til Slutt, fragmentene er PCR forsterket hvis det er nødvendig, og fragmenter er valgt størrelse (vanligvis ~300-500bp) for å avslutte bibliotek.
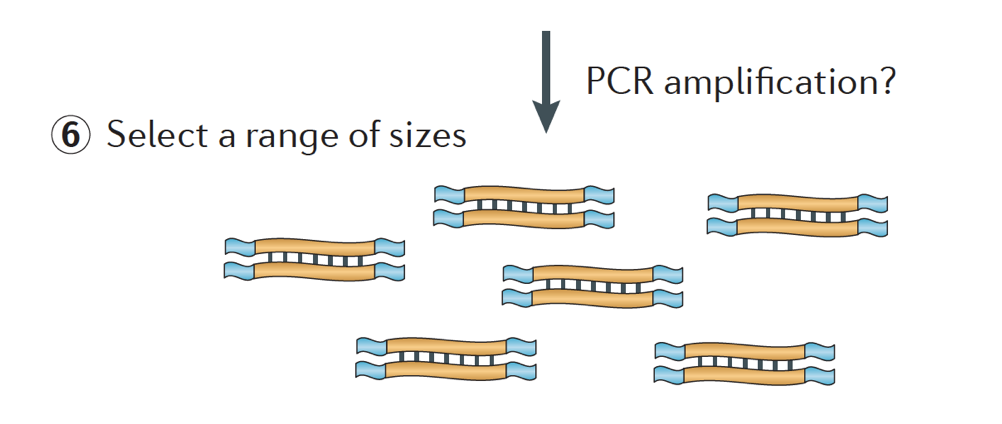
Bilde credit: Martin J. A. og Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina-Sekvensering
Enkelt-end versus Sammenkoblede-end
Etter forberedelse av biblioteker, sekvensering kan utføres for å generere nukleotide-sekvenser av endene av fragmenter, som er kalt leser. Du vil ha muligheten til sekvensering en enkelt slutten av cDNA fragment (enkelt-end leser) eller begge ender av fragmenter (sammenkoblede-end leser).,
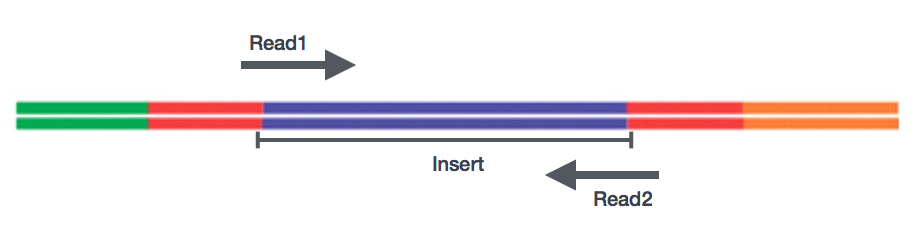
- SE – Enkelt slutten dataset => Bare Read1
- PE – Sammenkoblede-end dataset => Read1 + Read2
- kan være 2 separate FastQ filer eller bare en med interleaved par
Vanligvis enkelt-end-sekvensering er tilstrekkelig med mindre det forventes at den som leser vil matche flere steder på genom (f.eks. organismer med mange paralogous gener), sammenstillinger blir utført, eller for skjøte isoformen differensiering. Vær oppmerksom på at paret-end leser er generelt 2x dyrere.,
Forskjellige sekvensering plattformer
Det finnes en rekke Illumina plattformer å velge mellom å sekvens cDNA biblioteker.

Bilde credit: Tilpasset fra Illumina
Forskjeller i plattformen kan endre lengden på lyder generert, kvaliteten av lyder, samt det totale antall lesninger sekvensert per kjøre og den tiden som kreves for å sekvens bibliotekene., De forskjellige plattformene hver bruke en annen flyt celle, som er et glass overflate belagt med et arrangement av sammenkoblede oligos som er komplementære til kortene som er lagt til malen molekyler. Flyten celle er der sekvensering reaksjoner finner sted.

Bilde credit: Tilpasset fra Illumina
– Sekvensering-av-syntese
Illumina-sekvensering technology bruker en sekvensering-av-syntese tilnærming som er beskrevet i mer detalj nedenfor.
I trinn, DNA-fragmentene i cDNA bibliotek er denaturert og brukes til glass flow celle., Disse denaturert fragmenter binder seg til utfyllende oligos som allerede covalently bundet til flyten celle baner, noe som resulterer i vedlegg.
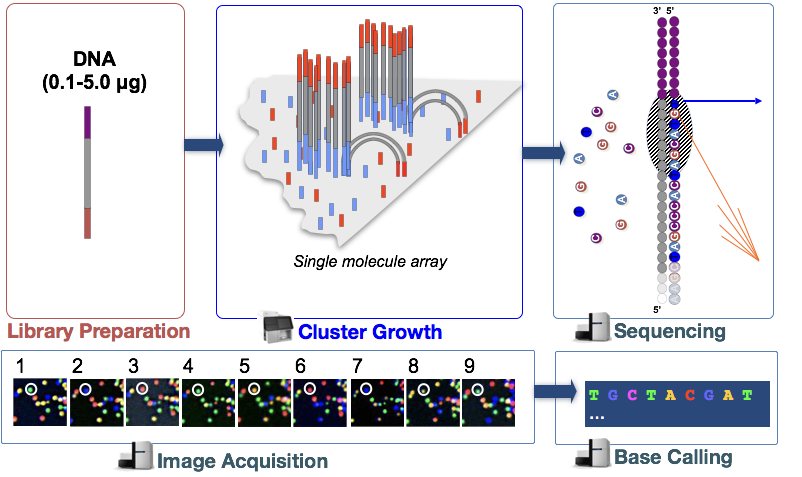
Klynge Generasjon
Når fragmenter har festet, en fase som kalles klynge generasjon begynner. Under dette trinnet, enkelt fragmenter er clonally forsterket for å skape en klynge (fragmenter i umiddelbar nærhet) av identiske fragmenter. Dette er nødvendig slik at fluorescens kan være lett tatt fra hver klynge, i stedet for et enkelt fragment, under nukleotid inkorporering i neste trinn.,
- Syntetisere den komplettere med polymerase
- dsDNA er denaturert, og opprinnelige DNA vasket bort etterlot syntetisert strand covalently bundet til å flyte celle.
- Enkelt strand hybridises med tilstøtende adapter for å danne en «bro’
- dsDNA er utvidet med polymerase. Hver strand covalently bundet til forskjellige adapter.
- Gjenta mange ganger for å clonally forsterke alle unike fragmenter på flyt celle for å danne klynger av samme rekkefølge.,
Sekvensering av syntese (& image acquisition)
Etter klynge generasjon, fluorescentmerkede-merket nukleotider inngår en om gangen (syklisk) og fluorescens bilder er tatt for å identifisere hvilke nukleotid blir innlemmet i hver klynge i hver syklus.
- Denaturering klynger og blokk 3′ endene til å hindre at uønskede priming.
- Hybridiserer sekvensering primere til adapteren rekkefølge på de løse endene.,
- Syklus fire NTPs med fluoriserende markører og terminator rekkefølge og polymerases.
- Når NTP er innarbeidet, klyngen er begeistret av en lyskilde og en karakteristisk fluroscent signalet avgis.
- farge er registrert, så terminator på dye er spaltet og vasket. Prosessen gjentas for specifioed antall sykluser.,
Base Ringer
Illumina har proprietær programvare som går gjennom alle bilder som er tatt i forrige fase, og genererer tekst filer med sekvensen informasjon om hvert cluster basert på fluorescens. I tillegg til å ringe baser, denne programvaren tildeler en probablity score for å indikere hvor sikker på at det var om å kalle noe en «A», en «T», en «G» eller en «C».
Hvis det er noen uklarheter, f.eks., i en viss syklus på bildet for en klynge som ikke har en bestemt farge som kan være assosiert med en bestemt nukleotid, base-telefoni programvare vil ha en lav sannsynlighet forbundet med det, og vil tilordne en «N» i stedet for «A», «T», «G» eller «C».
til slutt,
- Antall klynger ~= Antall lesninger
- Antall sekvensering sykluser = Lengde av lyder
antall sykluser (lengde leser) vil avhenge sekvensering plattformen som brukes så vel som dine preferanser.
MERK., Hvis du ønsker å utforske sekvensering av syntese mer i dybden, anbefaler vi at dette virkelig fin animasjon tilgjengelig på Illumina ‘ s YouTube-kanal.
Multiplexing
Avhengig av Illumina-plattformen (MiSeq, HiSeq, NextSeq), antall kjørefelt per flow celle, og antall lesninger som kan oppnås per kjørefelt varierer mye. Du må bestemme hvor mange leser du ønsker per sample (dvs. sequencning dybde) og da basert på plattform du velger å beregne hvor mange sum baner du vil kreve for sett med prøver., Vi vil snakke mer om hensynene når du gjør dette vedtaket i neste leksjon på Eksperimentell Hensyn
Vanligvis, gebyrer for sekvensering er per kjørefelt av flyten celle, og du vil være i stand til å kjøre flere prøver per kjørefelt. Illumina har derfor utviklet en fin sammensatt metode som lar biblioteker fra flere prøver å være samlet og sekvensert samtidig i samme kjørefelt på en flyt celle. Denne metoden krever tillegg av indekser (i Illumina-adapter) eller spesielle strekkoder (utenfor Illumina-adapter) som beskrevet i skjema nedenfor.,
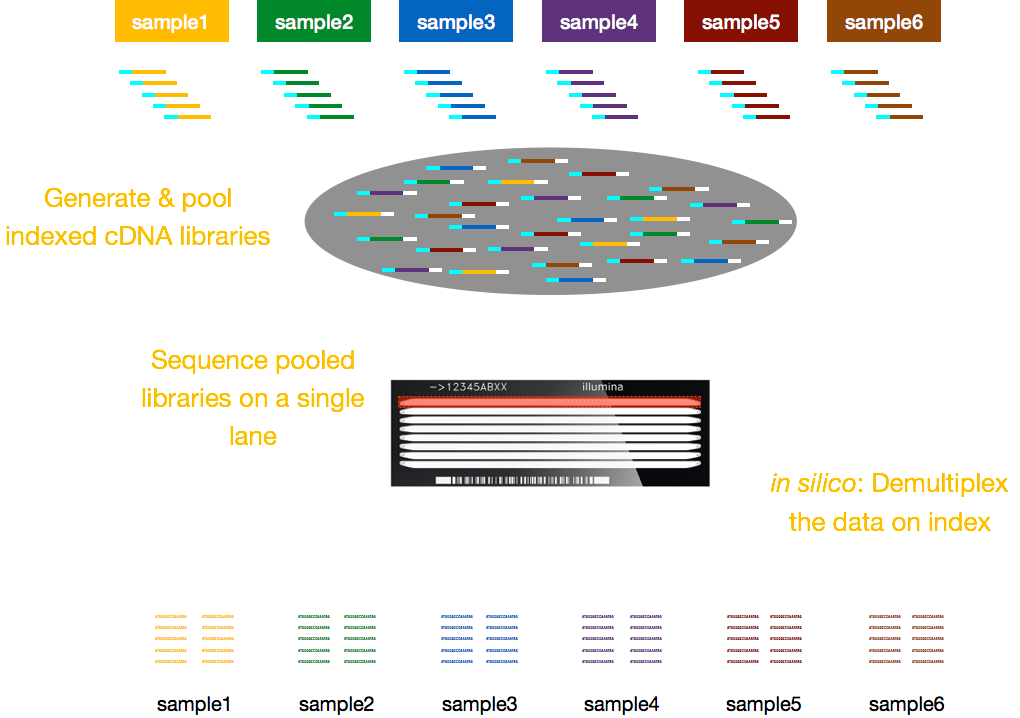
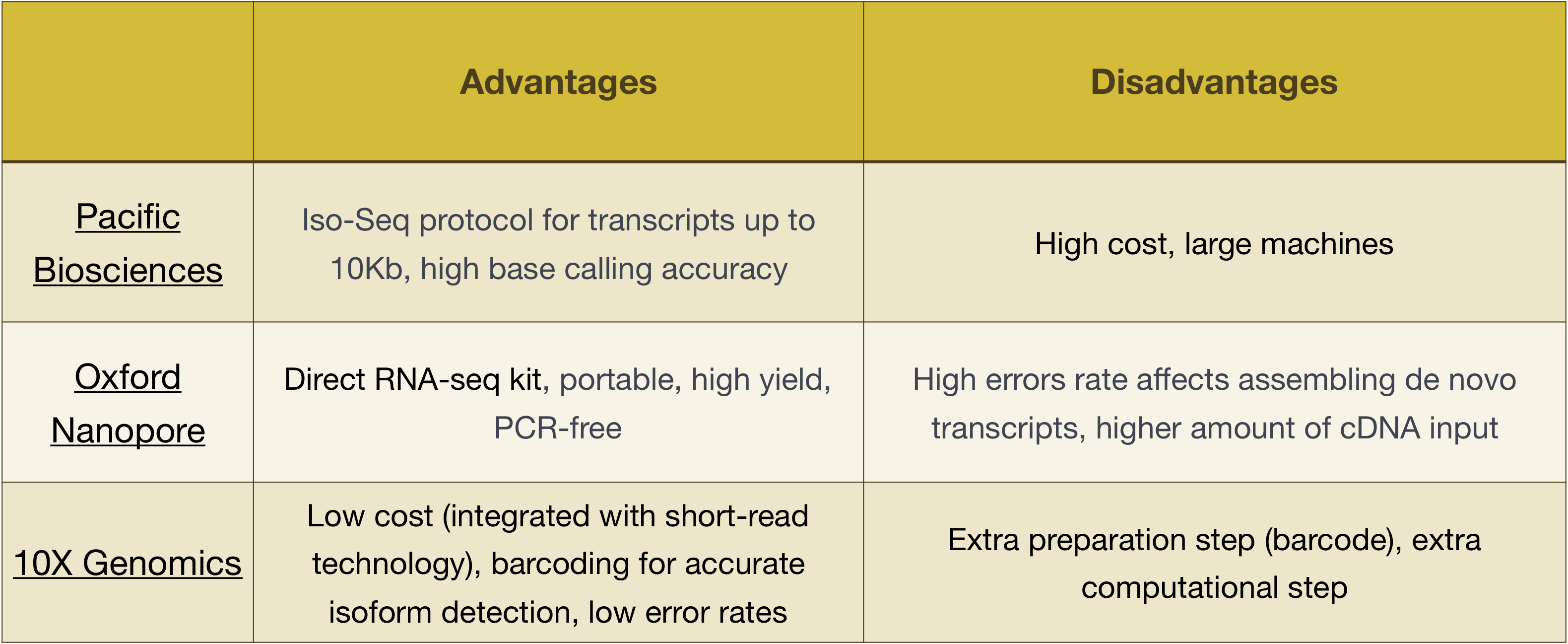
MERK: arbeidsflyten presentert i denne leksjonen er bestemt til Illumina-sekvensering, som er i dag den mest anvendte sekvensering metode., Men det er andre lange-les-sekvensering metoder verdt å merke seg, for eksempel:
- Pacific Biovitenskap: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomics: https://www.10xgenomics.com/
Fordeler og ulemper med disse teknologiene kan utforskes i tabellen nedenfor:
Legg igjen en kommentar