geschatte tijd: 90 minuten
leerdoelen:
- beschrijf het proces van RNA-seq library voorbereiding
- Beschrijf de Illumina sequencing methode
Inleiding tot RNA-seq
RNA-seq is een spannende experimentele techniek die gebruikt om genexpressie binnen of tussen voorwaarden te onderzoeken en/of te kwantificeren.,
zoals we weten, geven genen instructies om eiwitten aan te maken, die een bepaalde functie binnen de cel vervullen. Hoewel alle cellen dezelfde DNA-sequentie bevatten, zijn de spiercellen verschillend van zenuwcellen en andere types van cellen wegens de verschillende genen die in deze cellen en de verschillende geproduceerde RNAs en proteã nen worden aangezet.
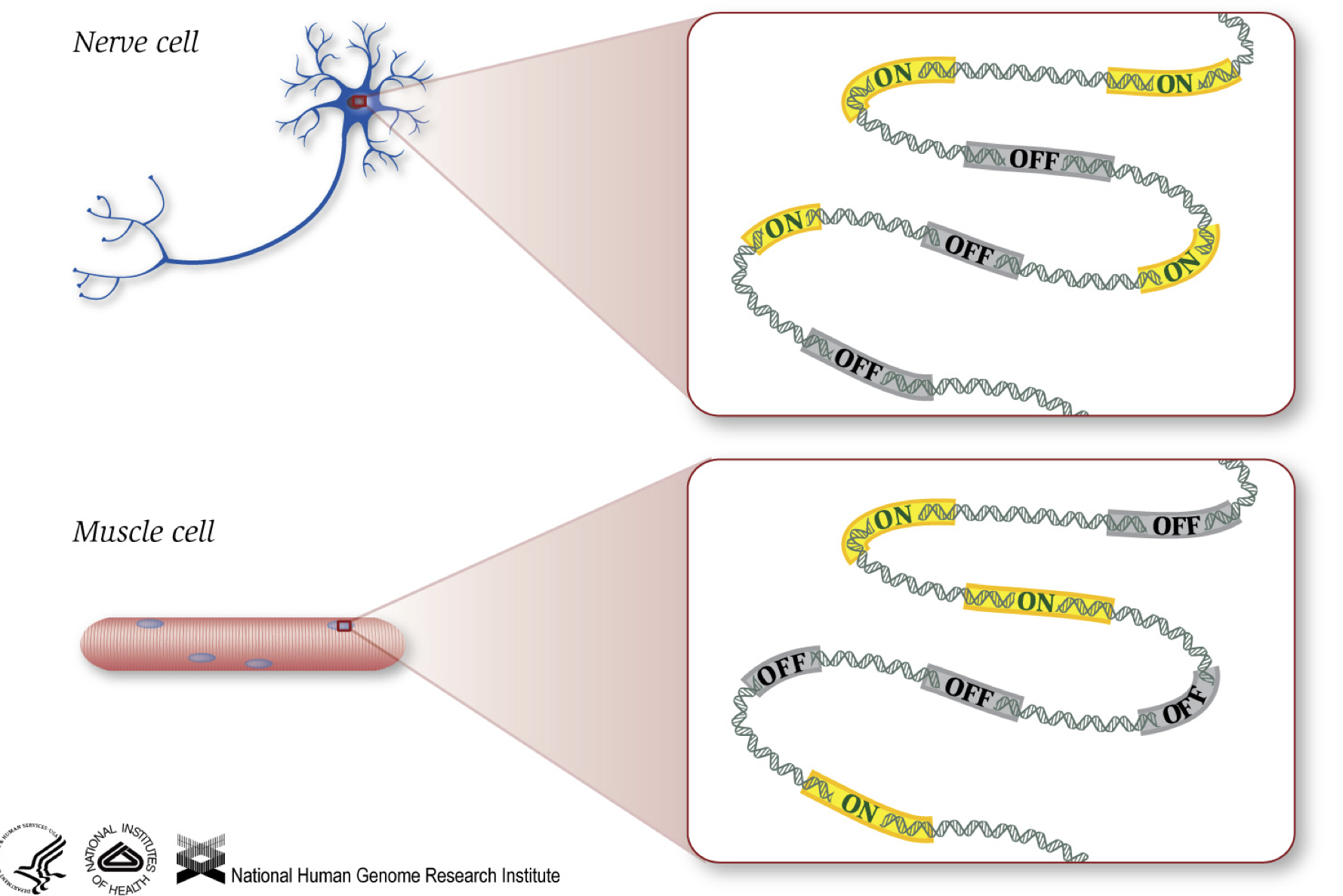
verschillende biologische processen, evenals mutaties, kunnen invloed hebben op welke genen worden ingeschakeld en welke worden uitgeschakeld, naast de hoeveelheid specifieke genen die aan / uit worden gezet.,
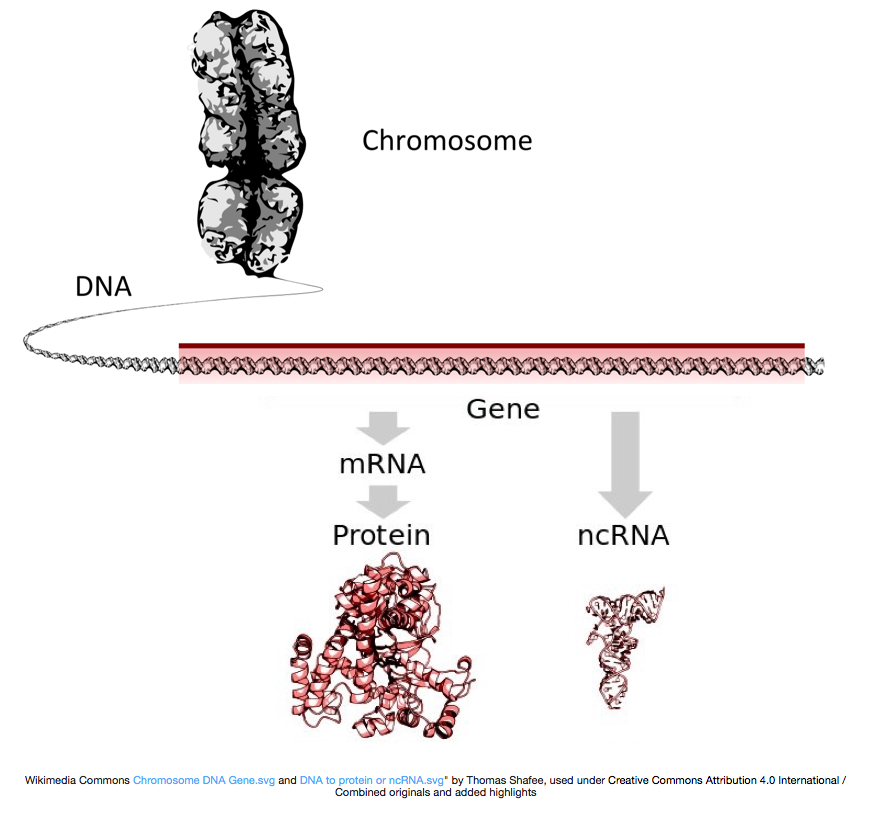
om eiwitten te maken, wordt het DNA getranscribeerd in boodschapper-RNA, of mRNA, dat door het ribosoom wordt vertaald in eiwitten. Nochtans, coderen sommige genen RNA dat niet in proteã ne wordt vertaald; Deze RNAs worden genoemd niet-codeert RNAs, of ncRNAs. Vaak hebben deze RNAs een functie in en van zichzelf en omvatten rRNAs, tRNAs, en siRNAs, onder anderen. Al RNAs getranscribeerd van genen worden genoemd transcripten.
om in eiwitten te worden vertaald, moet het RNA worden verwerkt om mRNA te genereren., In de figuur hieronder, vertegenwoordigt de hoogste bundel in het beeld een gen in DNA, samengesteld uit de onvertaalde gebieden (UTRs) en het open gelezen kader. De genen worden getranscribeerd in pre-mRNA, die nog de intronische opeenvolgingen bevat. Na post-transciptional verwerking, worden introns verbonden uit en een Polya staart en 5 ‘ GLB worden toegevoegd aan opbrengst Rijpe mRNA afschriften, die in proteã nen kunnen worden vertaald.,
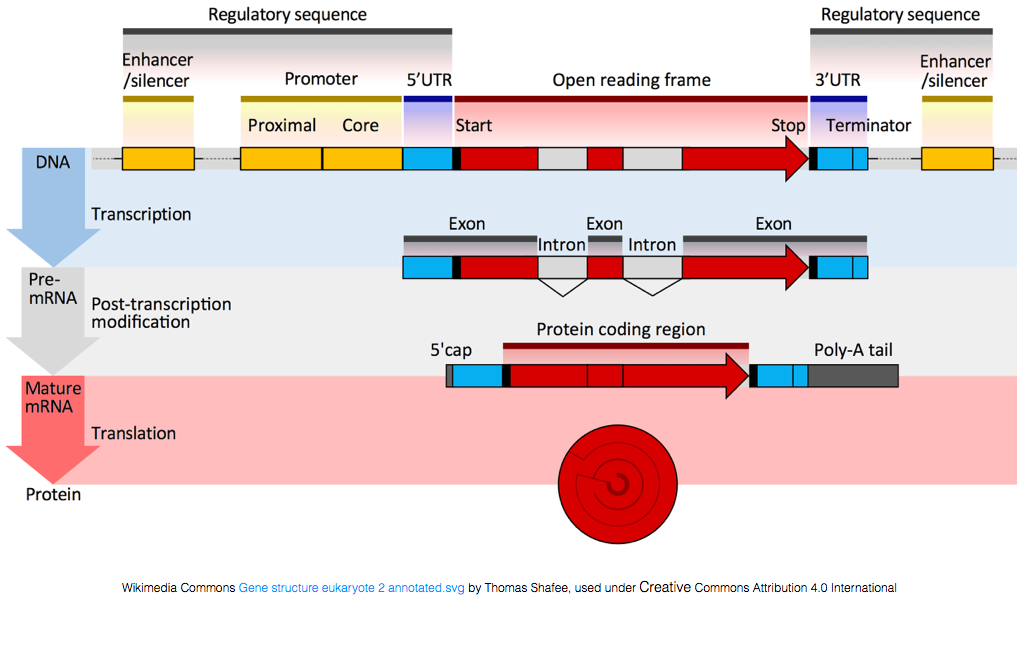
hoewel mRNA-transcripten een polyA-staart hebben, doen veel van de niet-coderende RNA-transcripten dat niet omdat de post-transcriptionele verwerking voor deze transcripten anders is.
Transcriptomics
het transcriptoom wordt gedefinieerd als een verzameling van alle transcript-uitlezingen in een cel., RNA-seq-gegevens kunnen worden gebruikt om het transcriptoom van een organisme te onderzoeken en/of te kwantificeren, dat kan worden gebruikt voor de volgende soorten experimenten:
- differentiële genexpressie: kwantitatieve evaluatie en vergelijking van transcriptniveaus
- Transcriptoomassemblage: opbouw van het profiel van getranscribeerde gebieden van het genoom, een kwalitatieve evaluatie.,
- kan worden gebruikt om betere genmodellen te bouwen en deze te verifiëren met behulp van de assemblage
- metatranscriptomics of communautaire transcriptoomanalyse
Illumina library preparation
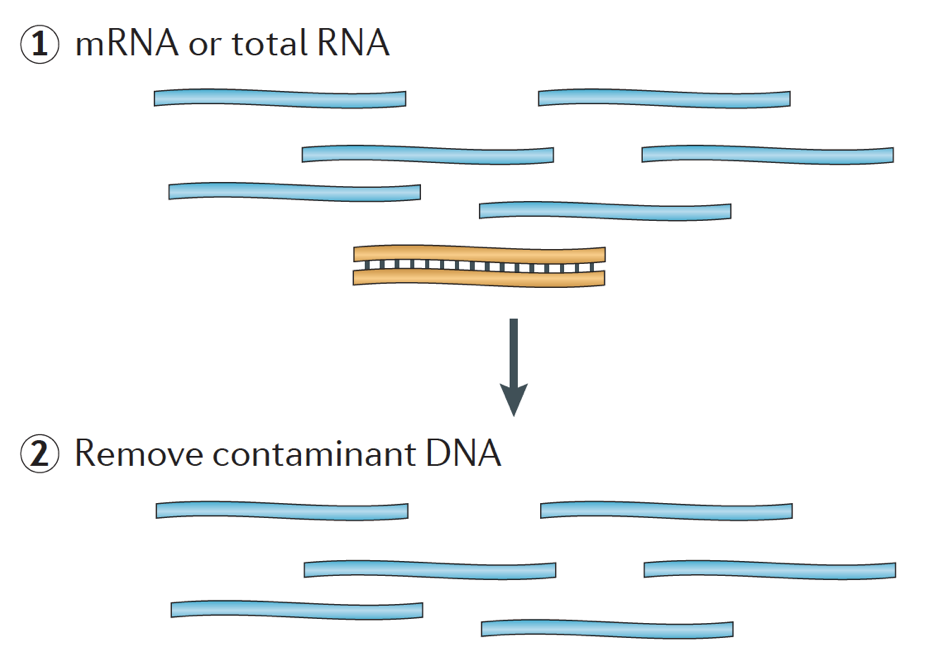
bij het starten van een RNA-seq-experiment moet voor elk monster het RNA worden geà soleerd en omgezet in een cDNA-bibliotheek voor sequencing. De algemene workflow voor bibliotheekvoorbereiding wordt beschreven in de onderstaande stap-voor-stap afbeeldingen.
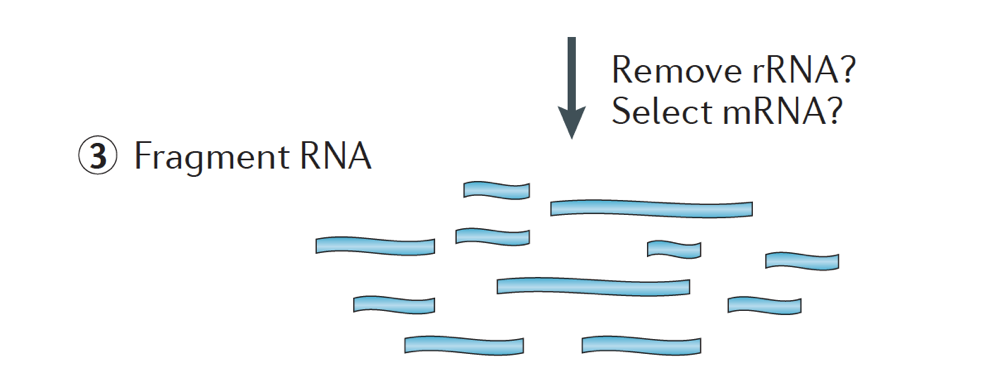
kort, RNA wordt geïsoleerd uit het monster en contaminerend DNA wordt verwijderd met DNase.,
het RNA-Monster ondergaat dan ofwel selectie van de mRNA (polyA-selectie) ofwel depletie van de rRNA. Het resulterende RNA is gefragmenteerd.
over het algemeen vertegenwoordigt ribosomaal RNA de meerderheid van de RNA ’s die aanwezig zijn in een cel, terwijl boodschapper-RNA’ s een klein percentage van totaal RNA vertegenwoordigen, ~2% in mensen. Daarom, als wij de eiwit-coderende genen willen bestuderen, moeten wij voor mRNA verrijken of rRNA uitputten., Voor differentiële genuitdrukkingsanalyse, is het het beste om voor Poly(A)+ te verrijken, tenzij u informatie over lange niet-codeert RNAs probeert te verkrijgen, dan een ribosomale depletie van RNA doet.
De grootte van de doelfragmenten in de uiteindelijke bibliotheek is een belangrijke parameter voor bibliotheekconstructie. De fragmentatie van DNA wordt typisch gedaan door fysische methodes (d.w.z., akoestisch scheren en sonication) of enzymatische methodes (d.w.z., niet-specifieke endonuclease cocktails en transposase tagmentation reacties.,
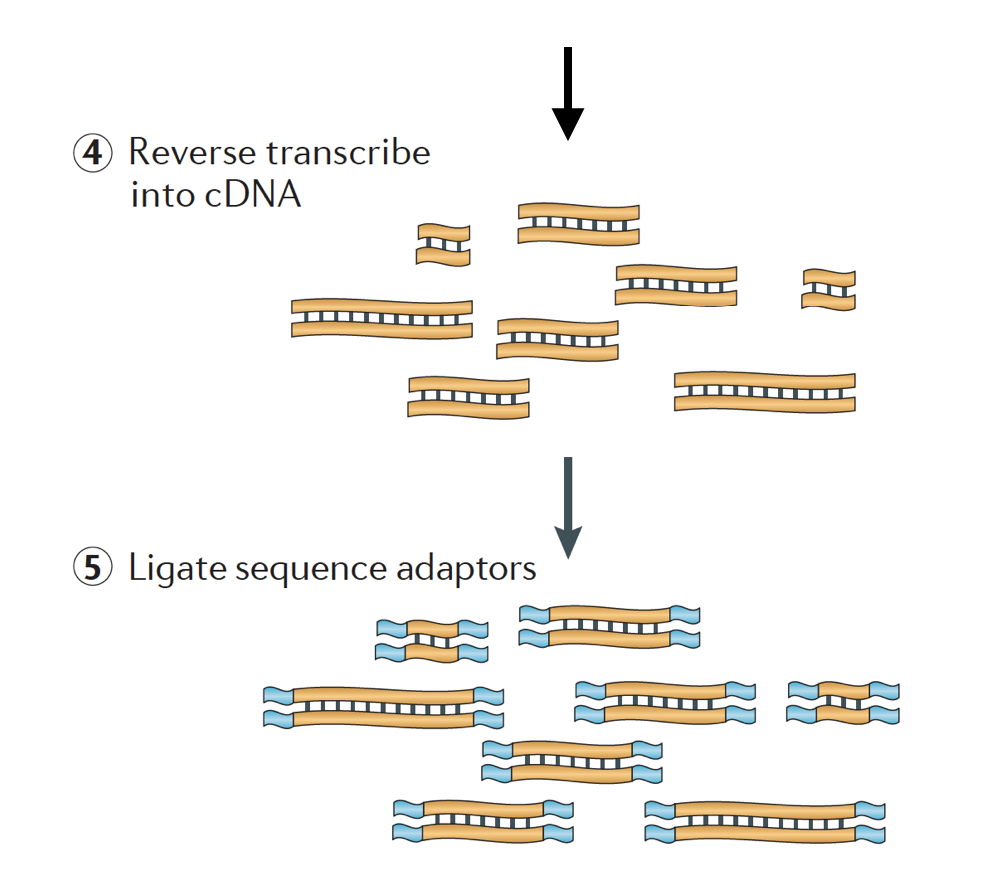
het RNA wordt dan omgekeerd getranscribeerd in dubbelstrengs cDNA en sequentieadapters worden vervolgens toegevoegd aan de uiteinden van de fragmenten.
de cDNA-bibliotheken kunnen op een manier worden gegenereerd om informatie te behouden over de DNA-streng waarvan het RNA is getranscribeerd. Bibliotheken die deze informatie behouden worden genoemd vastgelopen bibliotheken, die nu standaard met truseq van Illumina vastgelopen RNA-Seq uitrustingen zijn., Stranded libraries zouden niet duurder moeten zijn dan unstranded, dus er is eigenlijk geen reden om deze aanvullende informatie niet aan te schaffen.,
Er zijn 3 soorten gen bibliotheken beschikbaar:
- Doorsturen (secondstrand) – leest lijken op het gen sequentie of de secondstrand gen sequentie
- Omkeren (firststrand) – leest lijken op de aanvulling van het gen sequentie of firststrand gen sequentie (TruSeq)
- Unstranded
tot slot, de fragmenten zijn PCR-geamplificeerde als dat nodig is, en de fragmenten zijn grootte is geselecteerd (meestal ~300-500bp) voor de afwerking van de bibliotheek.
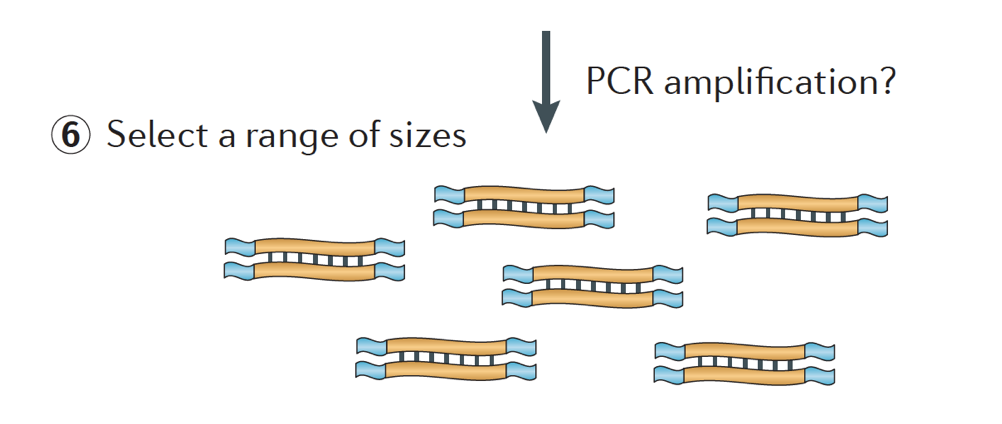
beeld door: Martin J. A. and Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina Sequencing
Single-end versus Paired-end
na voorbereiding van de bibliotheken kan sequencing worden uitgevoerd om de nucleotidesequenties van de uiteinden van de fragmenten te genereren, die reads worden genoemd. U zult de keus hebben van het rangschikken van één enkel eind van de cDNA fragmenten (single-end leest) of beide einden van de fragmenten (paired-end leest).,
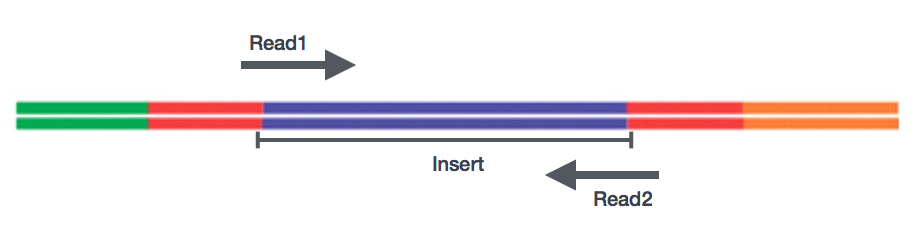
- SE – Single einde dataset => Alleen Read1
- PE – Paired-end dataset => Read1 + Lezen2
- kan worden 2 aparte FastQ bestanden of gewoon een met interleaved paren
over het Algemeen één-einde-sequencing is voldoende, tenzij verwacht wordt dat de leest zal overeenkomen met meerdere locaties op het genoom (bijv. organismen met veel paralogous genen), assembly ‘ s worden uitgevoerd, of voor splice isovorm differentiatie. Wees ervan bewust dat gepaarde-einde leest zijn over het algemeen 2x duurder.,
verschillende sequentieplatforms
Er zijn verschillende platforms van Illumina om uit te kiezen om de cDNA-bibliotheken te sequencen.

Image credit: aangepast van Illumina
verschillen in platform kunnen de lengte van gegenereerde reads, de kwaliteit van reads, evenals het totale aantal reads sequenced per run en de hoeveelheid tijd die nodig is om de bibliotheken te sequencen veranderen., De verschillende platforms gebruiken elk een andere stroomcel, een glasoppervlak dat is gecoat met een opstelling van gepaarde oligos die complementair zijn aan de adapters die aan uw sjabloonmoleculen zijn toegevoegd. De stroomcel is waar de het rangschikken reacties plaatsvinden.

Beeldkrediet: aangepast van Illumina
Sequencing-by-synthesis
Illumina sequencing technology maakt gebruik van een sequencing-by-synthesis benadering die hieronder nader wordt beschreven.
In de stap worden de DNA-fragmenten in de cDNA-bibliotheek gedenatureerd en op de glasstroomcel aangebracht., Deze gedenatureerde fragmenten binden aan de complementaire oligos die reeds covalent gebonden zijn aan de stromingscelwegen, resulterend in gehechtheid.
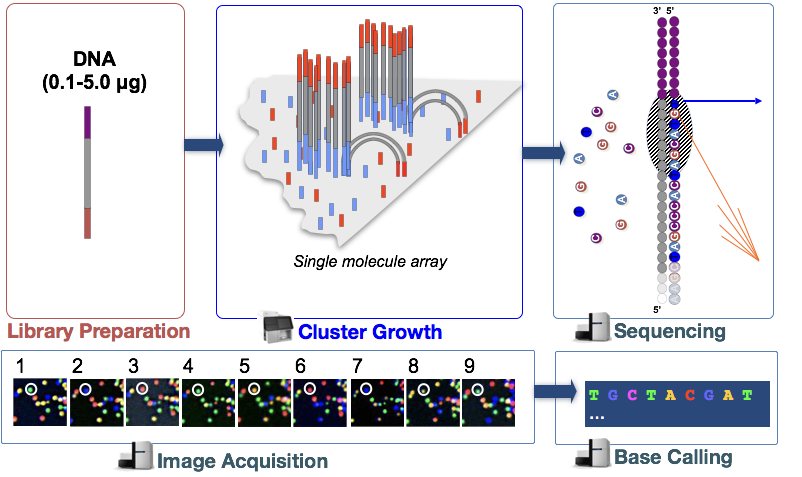
Clustergeneratie
zodra de fragmenten zijn aangesloten, begint een fase genaamd clustergeneratie. Tijdens deze stap, worden de enige fragmenten clonally versterkt om tot een cluster (fragmenten in dichte nabijheid) van identieke fragmenten te leiden. Dit is noodzakelijk zodat de fluorescentie gemakkelijk van elke cluster, in plaats van één enkel fragment, tijdens nucleotideinbouw in de volgende stap kan worden gevangen.,
- synthetiseer het complement met polymerase
- dsDNA wordt gedenatureerd, en origineel DNA wordt weggespoeld, waardoor gesynthetiseerde streng covalent gebonden is aan stroomcel.
- single strand hybridiseert met aangrenzende adapter om een ‘bridge’te vormen
- dsDNA wordt uitgebreid met polymerase. Elke streng covalent gebonden aan verschillende adapter.
- herhaal vele malen om alle unieke fragmenten op stroomcel clonaal te versterken om clusters van identieke opeenvolging te vormen.,
Sequencing door synthese (& beeldopname)
Na clustergeneratie worden één voor één (cyclisch) fluorescentiebeelden opgenomen om te bepalen welk nucleotide in elke cluster in elke cyclus wordt opgenomen.
- Denatureclusters en blok 3′ eindigt om ongewenste priming te voorkomen.
- Hybrideer sequentieprimers naar adaptersequentie aan de losse eindjes.,
- cyclus vier NTP ‘ s met fluorescerende markers en terminatorsequentie en polymerasen.
- zodra NTP is opgenomen, wordt het cluster opgewekt door een lichtbron en wordt een karakteristiek fluroscentsignaal uitgezonden.
- de kleur wordt geregistreerd, dan wordt de terminator op kleurstof gesplitst en gewassen. Proces herhaalt voor specifiek aantal cycli.,
Basisaanroep
Illumina heeft propriëtaire software die door alle beelden gaat die in de vorige fase zijn vastgelegd en tekstbestanden genereert met sequentieinformatie over elk cluster op basis van de fluorescentie. Naast het aanroepen van de bases, kent deze software een waarschijnlijkheidsscore toe om aan te geven hoe zeker het was over het aanroepen van iets een “A”, een “T”, een “G” of een “C”.
als er onduidelijkheden zijn, bijv., bij een bepaalde cyclus heeft het beeld voor een cluster geen verschillende kleur die met een specifiek nucleotide kan worden geassocieerd, zal de basis roepende software een lage waarschijnlijkheid verbonden met het hebben en zou een “N” in plaats van “A”, “T”, “G” of “C”toewijzen.
ter afsluiting,
- aantal clusters ~= Aantal reads
- Aantal sequentiecycli = lengte van reads
het aantal cycli (lengte van de reads) zal afhangen van het gebruikte sequentieplatform en uw voorkeuren.
opmerking., Als je sequencing door synthese in meer diepte wilt verkennen, raden we deze echt leuke animatie aan die beschikbaar is op Illumina ‘ s YouTube-kanaal.
Multiplexing
afhankelijk van het Illumina-platform (MiSeq, HiSeq, NextSeq), varieert het aantal rijstroken per stroomcel en het aantal meetwaarden dat per rijstrook kan worden verkregen sterk. U moet beslissen hoeveel reads u wilt per sample (dat wil zeggen de sequencning diepte) en vervolgens op basis van het platform dat u kiest berekenen hoeveel totale rijstroken u nodig hebt voor uw set van samples., We zullen meer praten over overwegingen bij het maken van deze beslissing in de volgende les over experimentele overwegingen
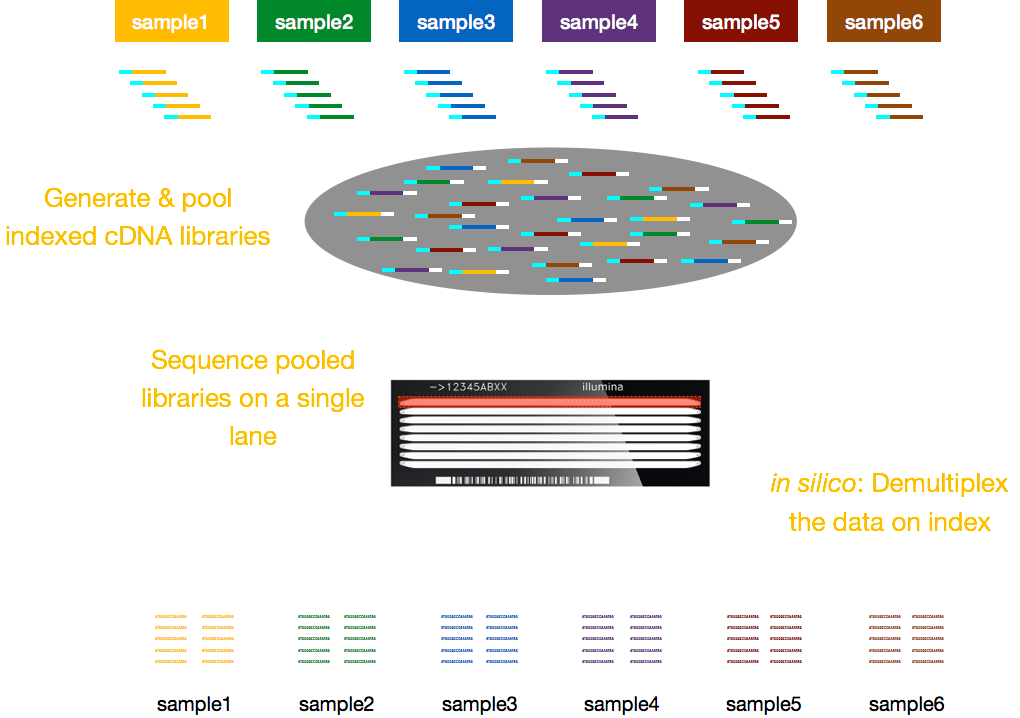
normaal gesproken zijn de kosten voor sequencing per rijstrook van de stroomcel en kunt u meerdere monsters per rijstrook uitvoeren. Illumina heeft daarom een mooie multiplexmethode ontwikkeld die het mogelijk maakt dat bibliotheken van verschillende monsters gelijktijdig worden samengevoegd en gesequenced in dezelfde rijstrook van een stroomcel. Deze methode vereist de toevoeging van indices (binnen de Illumina adapter) of speciale barcodes (buiten de Illumina adapter) zoals beschreven in het onderstaande schema.,
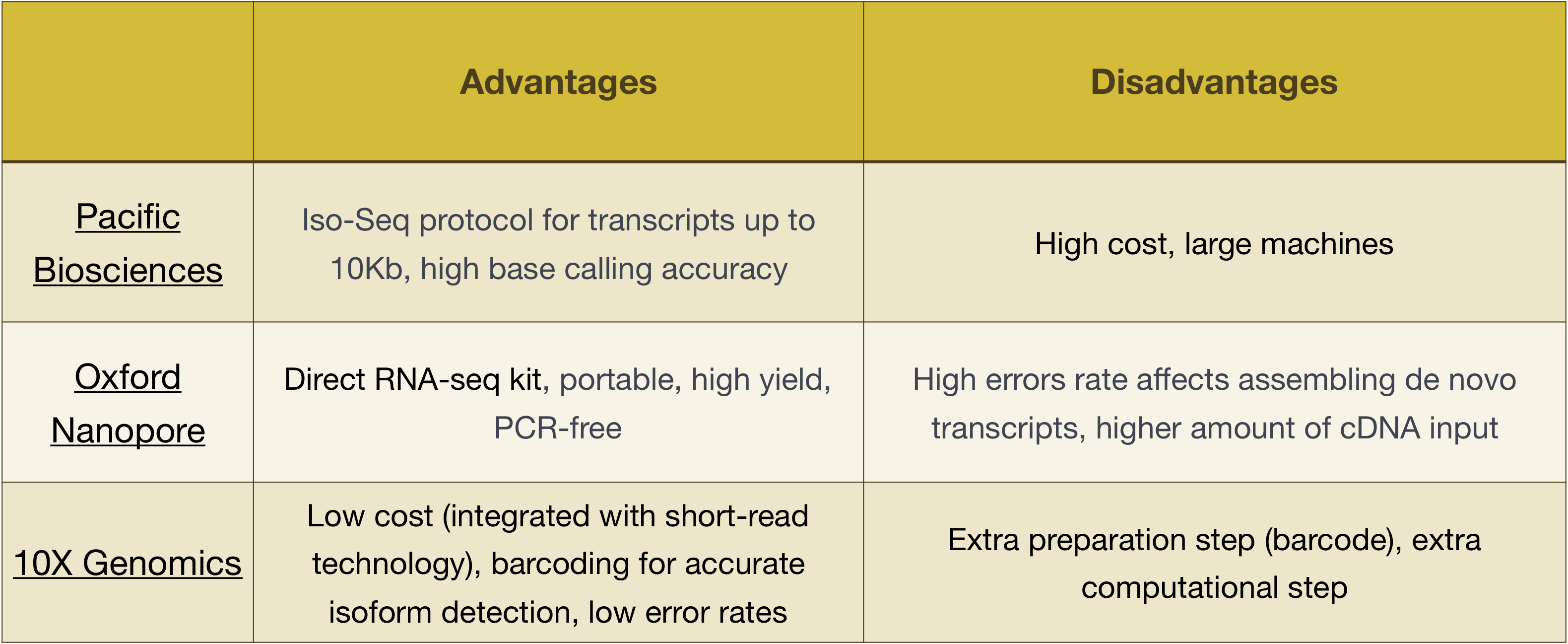
opmerking: de workflow in deze les is specifiek voor Illumina sequencing, wat momenteel de meest gebruikte sequencing methode is., Maar er zijn andere lange-lees sequencing methoden vermeldenswaard, zoals:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomics: https://www.10xgenomics.com/
Voordelen en nadelen van deze technologieën kan worden verkend in de tabel hieronder:
Geef een reactie