przybliżony czas: 90 minut
cele uczenia się:
- opisz proces przygotowania biblioteki RNA-seq
- opisz metodę sekwencjonowania Illumina
Wprowadzenie do RNA-seq
RNA-seq to ekscytująca metoda, która pozwala na Technika eksperymentalna, która jest wykorzystywana do zbadania i/lub kwantyfikacji ekspresji genów w obrębie lub między Warunkami.,
jak wiemy, geny dostarczają instrukcji tworzenia białek, które pełnią pewną funkcję w komórce. Chociaż wszystkie komórki zawierają tę samą sekwencję DNA, komórki mięśniowe różnią się od komórek nerwowych i innych typów komórek ze względu na różne geny, które są włączone w tych komórkach i różnych RNA i białek produkowanych.
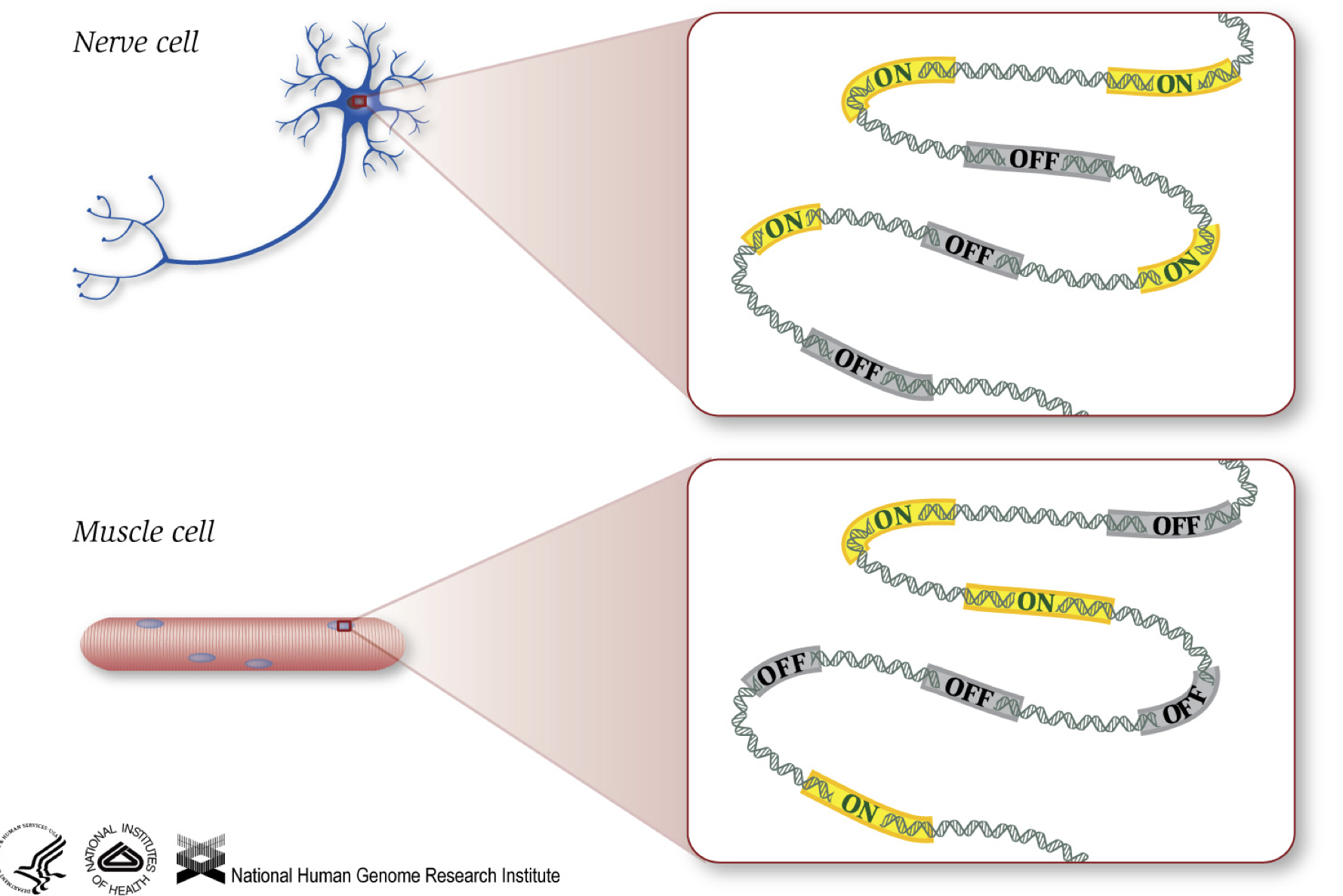
różne procesy biologiczne, a także mutacje, mogą mieć wpływ na to, które geny są włączone, a które wyłączone, oprócz tego, ile specyficznych genów jest włączonych / wyłączonych.,
aby wytworzyć białka, DNA jest transkrybowane do messenger RNA, lub mRNA, który jest tłumaczony przez rybosom do białka. Jednak niektóre geny kodują RNA, które nie zostaje przetłumaczone na białko; te RNA nazywane są niekodującymi RNA lub ncrna. Często te RNA mają funkcję w i od siebie i obejmują rRNA, tRNA i siRNAs, między innymi. Wszystkie RNA transkrybowane z genów nazywane są transkryptami.
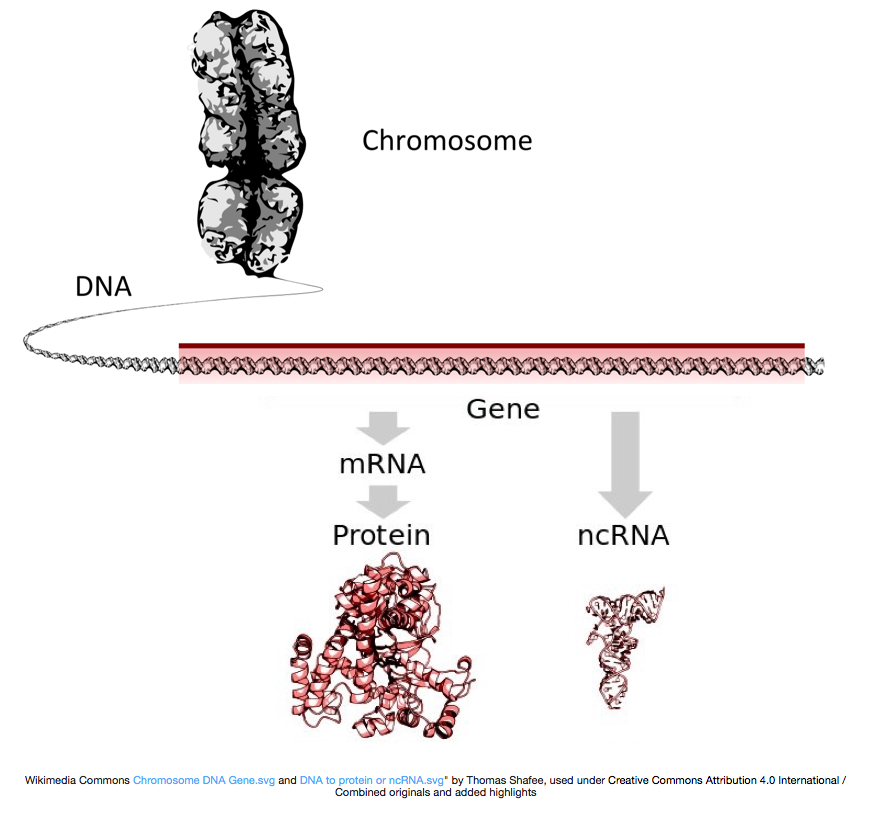
aby przekształcić się w białka, RNA musi zostać poddane obróbce w celu wytworzenia mRNA., Na poniższym rysunku górna nić obrazu reprezentuje gen w DNA, składający się z nieprzetłumaczonych regionów (UTRs) i otwartej ramki odczytu. Geny są transkrybowane do pre-mRNA, który nadal zawiera sekwencje introniczne. Po przetworzeniu post-transciptional introny są splatane i dodaje się polia tail i 5 ' cap w celu uzyskania dojrzałych transkryptów mRNA, które mogą być przekształcone w białka.,
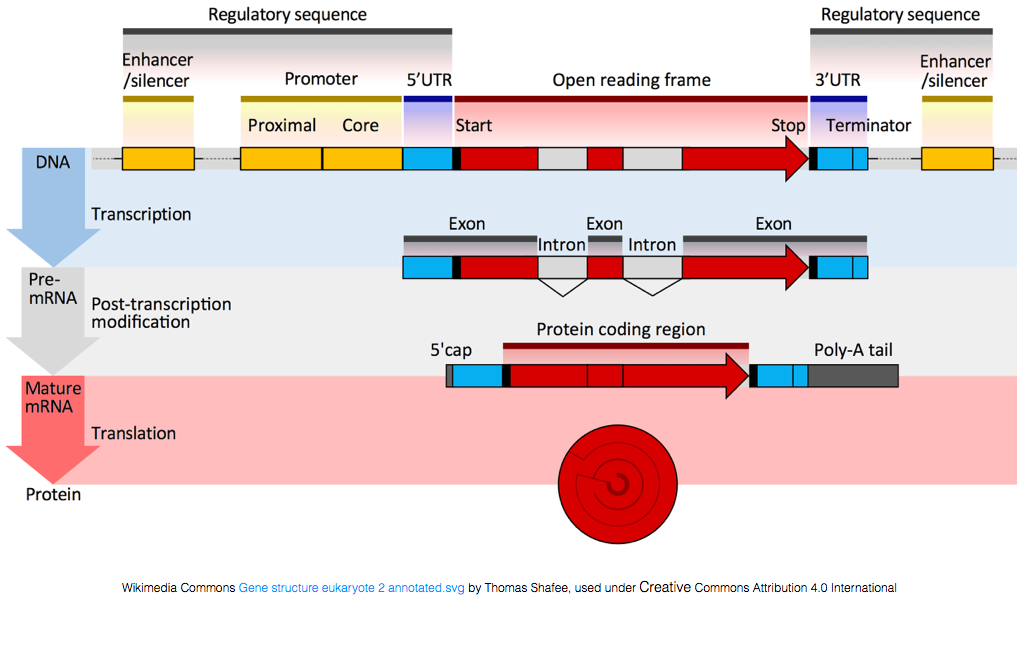
podczas gdy transkrypty mRNA mają ogon polia, wiele niekodujących transkryptów RNA nie ma, ponieważ przetwarzanie post-transkrypcyjne jest INNE dla tych transkryptów.
Transkryptomika
transkryptom jest zdefiniowany jako zbiór wszystkich odczytów transkryptu obecnych w komórce., Dane RNA-seq mogą być wykorzystane do badania i / lub kwantyfikacji transkryptomu organizmu, które mogą być wykorzystane do następujących rodzajów eksperymentów:
- różnicowa ekspresja genów: ocena ilościowa i porównanie poziomów transkryptu
- zespół transkryptomu: budowanie profilu transkrybowanych regionów genomu, ocena jakościowa.,
- może być użyty do pomocy w budowaniu lepszych modeli genów i ich weryfikacji przy użyciu assembly
- Metatranscriptomics lub community transcriptome analysis
przygotowanie biblioteki Illumina
podczas rozpoczynania eksperymentu RNA-seq, dla każdej próbki RNA musi być wyizolowany i przekształcony w bibliotekę cDNA do sekwencjonowania. Ogólny przebieg pracy nad przygotowaniem biblioteki jest opisany na poniższych ilustracjach krok po kroku.
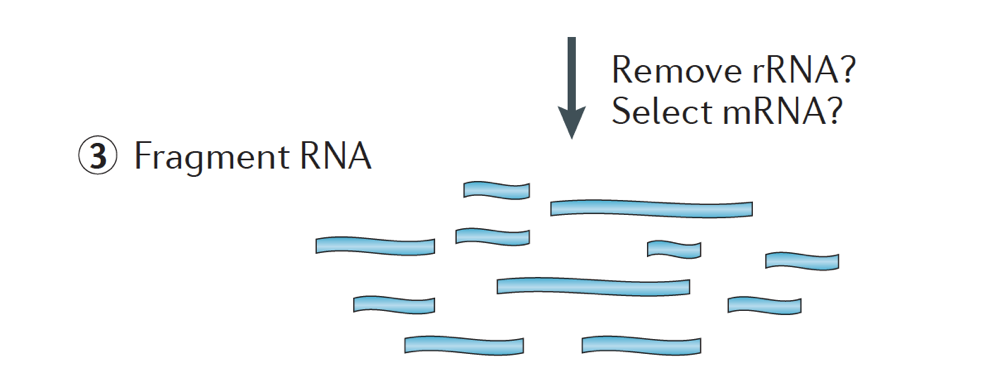
krótko mówiąc, RNA jest izolowany z próbki, a skażające DNA jest usuwane za pomocą DNazy.,
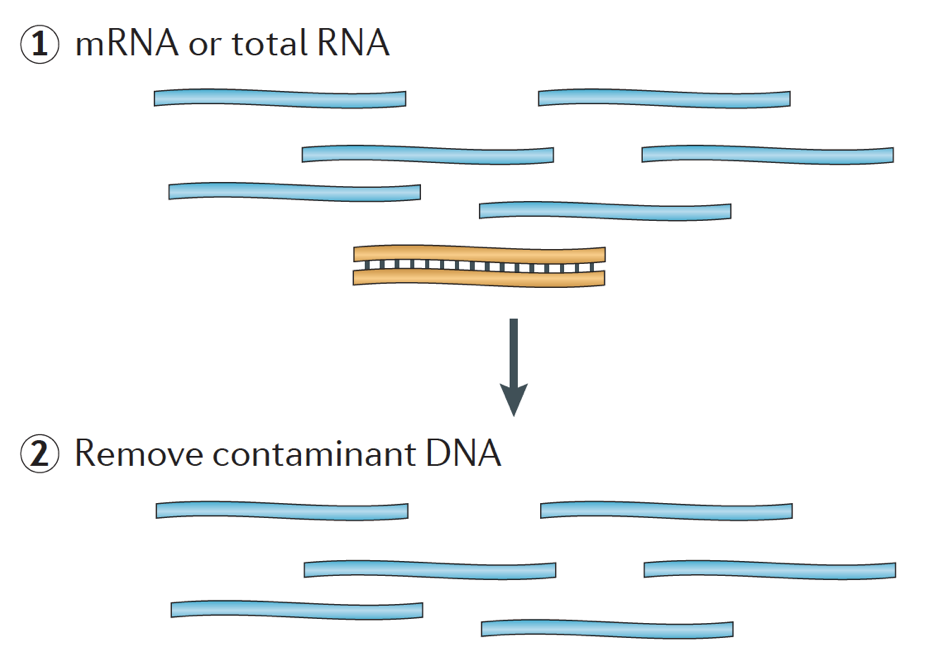
Próbka RNA poddawana jest następnie selekcji mRNA (polyA selection) lub wyczerpaniu rRNA. Powstałe RNA jest rozdrobnione.
ogólnie rzecz biorąc, rybosomalne RNA reprezentuje większość RNA obecnych w komórce, podczas gdy messenger RNA reprezentuje niewielki procent całkowitego RNA, ~2% u ludzi. Dlatego, jeśli chcemy badać geny kodujące białka, musimy wzbogacić się o mRNA lub zubożyć rRNA., W przypadku analizy ekspresji genów różnicowych najlepiej jest wzbogacić dla Poli (A)+, chyba że dążysz do uzyskania informacji o długich niekodujących RNA, a następnie zrób rybosomalne zubożenie RNA.
rozmiar docelowych fragmentów w ostatecznej bibliotece jest kluczowym parametrem dla budowy biblioteki. Fragmentacja DNA jest zwykle wykonywana metodami fizycznymi (tj. akustyczne ścinanie i sonikacja) lub metodami enzymatycznymi (tj. nieswoiste koktajle endonukleazowe i reakcje tagmentacji transpozazy.,
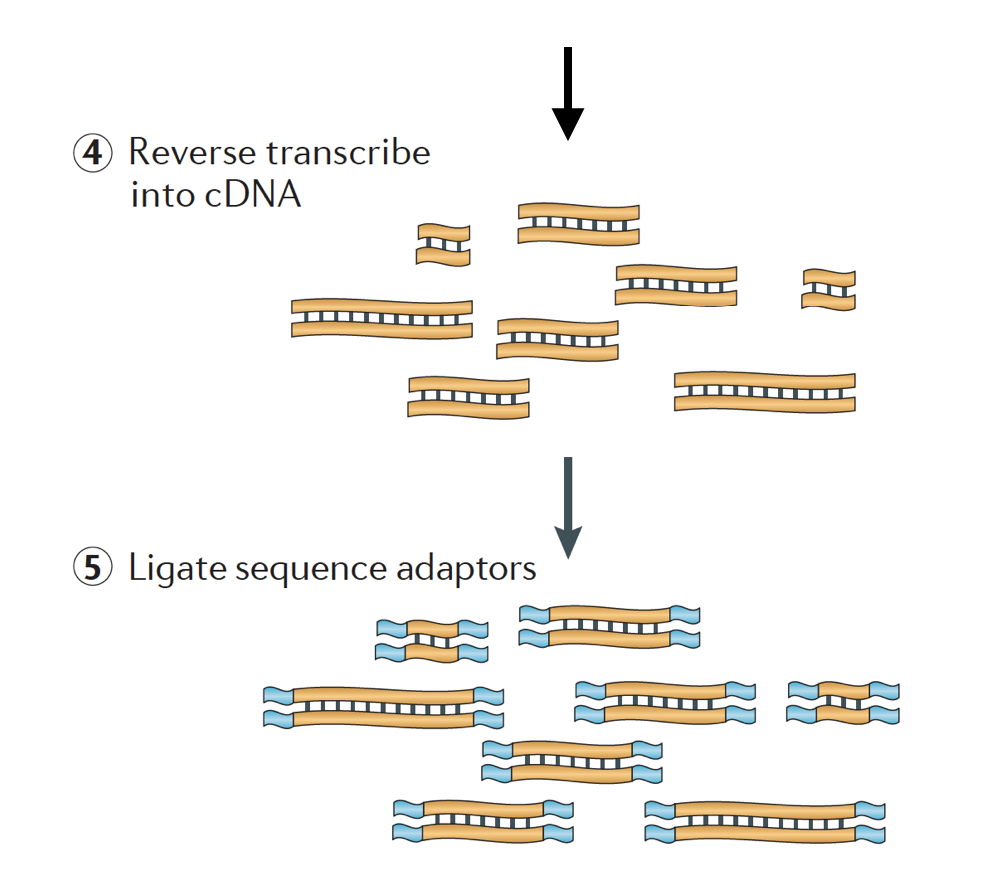
RNA jest następnie odwrotnie transkrybowany do dwuniciowych cDNA i Adaptery sekwencji są następnie dodawane do końców fragmentów.
biblioteki cDNA mogą być generowane w taki sposób, aby zachować informacje o tym, z której nici DNA został transkrybowany RNA. Biblioteki, które przechowują te informacje, nazywane są bibliotekami osieroconymi, które są obecnie standardem w zestawach truseq stranded RNA-Seq firmy Illumina., Biblioteki osierocone nie powinny być droższe od nierozwiązanych, więc nie ma powodu, aby nie nabywać tych dodatkowych informacji.,
dostępne są 3 typy bibliotek cDNA:
- Forward (secondstrand) – odczyty przypominają sekwencję genu lub secondstrand cDNA
- Reverse (firststrand) – odczyty przypominają dopełniacz sekwencji genu lub firststrand cDNA (TruSeq)
- Unstranded
wreszcie, fragmenty są wzmocnione PCR w razie potrzeby, a fragmenty są wybrane rozmiar (zwykle ~300-500bp), aby zakończyć bibliotekę.
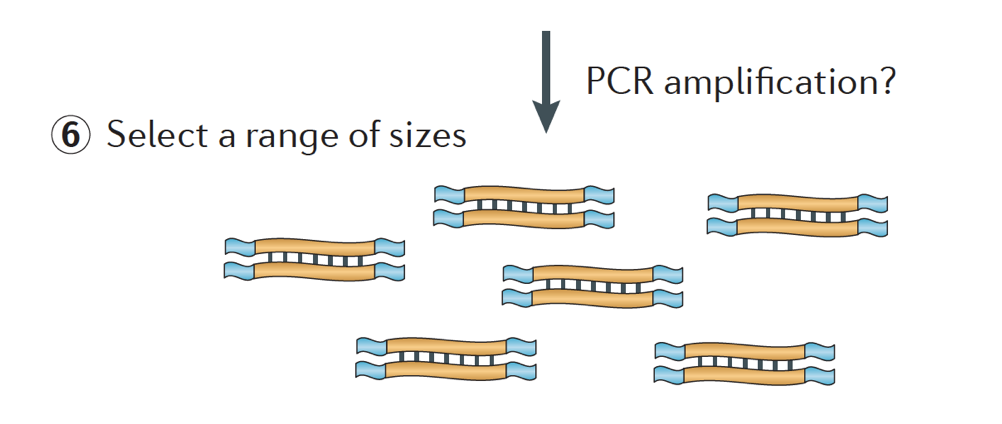
Image credit: Martin J. A. and Wang Z., Nat. Rev., Genet. (2011) 12:671-682
sekwencjonowanie Illumina
Single-end versus Paired-end
Po przygotowaniu bibliotek, sekwencjonowanie może być wykonywane w celu wygenerowania sekwencji nukleotydowych końców fragmentów, które nazywane są czytaniami. Będziesz miał do wyboru sekwencjonowanie pojedynczego końca fragmentów cDNA (odczyty pojedyncze) lub obu końców fragmentów (odczyty końcowe sparowane).,
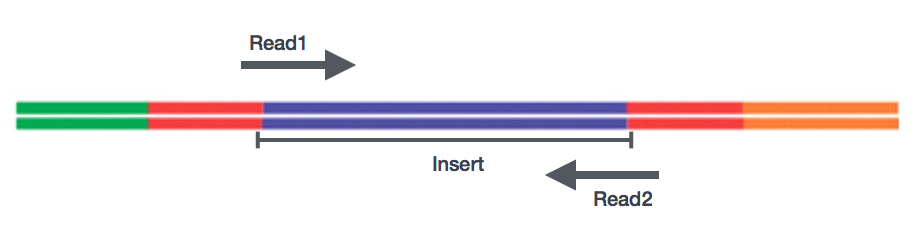
- SE – Single end dataset => only Read1
- PE – Paired-end dataset => Read1 + Read2
- can być 2 oddzielne pliki FASTQ lub tylko jeden z parami przeplatanymi
generalnie sekwencjonowanie jednokońcowe jest wystarczające, chyba że oczekuje się, że odczyty będą pasować do wielu lokalizacji w genomie (np. organizmy z wieloma paralogicznymi genami), zespoły są wykonywane lub dla różnicowania izoform splicingu. Należy pamiętać, że odczyty sparowane są na ogół 2x droższe.,
różne platformy sekwencjonowania
istnieje wiele platform Illumina do wyboru, aby uporządkować biblioteki cDNA.

kredyt obrazu: zaadaptowany z Illumina
różnice w platformie mogą zmieniać długość generowanych odczytów, jakość odczytów, a także całkowitą liczbę odczytów sekwencjonowanych na uruchomienie i czas potrzebny na uporządkowanie bibliotek., Każda z różnych platform wykorzystuje inną komórkę przepływową, która jest szklaną powierzchnią pokrytą układem sparowanych oligos, które uzupełniają Adaptery dodawane do cząsteczek szablonu. Komora przepływowa jest miejscem, w którym zachodzą reakcje sekwencjonowania.

kredyt obraz: zaadaptowany z Illumina
sekwencjonowanie przez syntezę
Technologia sekwencjonowania Illumina wykorzystuje podejście sekwencjonowanie przez syntezę, które jest opisane bardziej szczegółowo poniżej.
w tym kroku fragmenty DNA w bibliotece cDNA są denaturowane i nanoszone na szklaną komórkę przepływową., Te denaturowane fragmenty wiążą się z komplementarnymi oligosami, które są już kowalencyjnie związane z pasami komórek przepływowych, powodując ich przyłączenie.
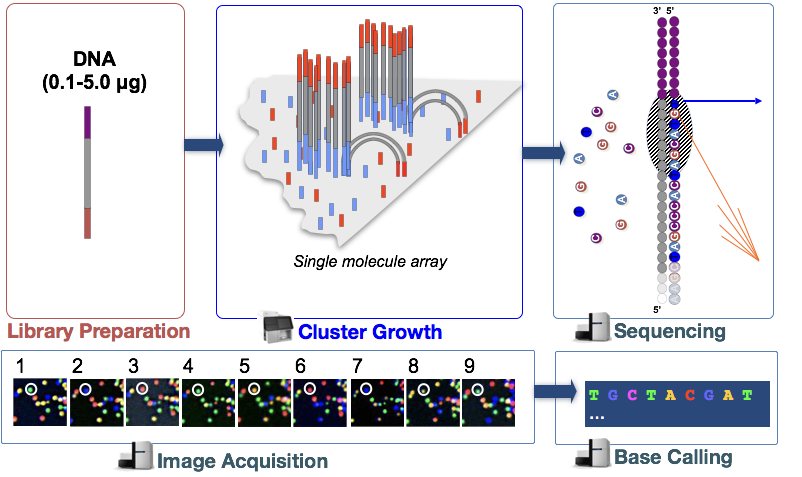
generowanie klastrów
Po dołączeniu fragmentów rozpoczyna się faza zwana generowaniem klastrów. Podczas tego etapu pojedyncze fragmenty są wzmacniane klonalnie, aby utworzyć klaster (fragmenty w bliskiej odległości) identycznych fragmentów. Jest to konieczne, aby fluorescencja mogła być łatwo wychwycona z każdego klastra, zamiast pojedynczego fragmentu, podczas inkorporacji nukleotydu w następnym kroku.,
- synteza dopełniacza za pomocą polimerazy
- dsDNA ulega denaturacji, a oryginalne DNA jest wymywane, pozostawiając zsyntetyzowaną nić kowalencyjnie związaną z komórką przepływową.
- jednożyłowa hybryda z sąsiednim adapterem w celu utworzenia „mostu”
- dsDNA jest przedłużona polimerazą. Każde pasmo kowalencyjnie związane z innym adapterem.
- powtarzaj wiele razy, aby clonalnie wzmocnić wszystkie unikalne fragmenty na komórce przepływowej, tworząc klastry identycznej sekwencji.,
sekwencjonowanie przez syntezę (& akwizycja obrazu)
po wygenerowaniu klastra fluorescencyjnie oznaczone nukleotydy są włączane pojedynczo (cyklicznie) i obrazy fluorescencji są rejestrowane w celu określenia, który nukleotyd zostaje włączony do każdego klastra w każdym cyklu.
- klastry Denaturowe i blok 3′ kończą się, aby zapobiec niepożądanemu gruntowaniu.
- ,
- cykl czwarty NTPs z fluorescencyjnymi markerami i sekwencją Terminatorów oraz polimerazami.
- Po włączeniu NTP klaster jest wzbudzany przez źródło światła i emitowany jest charakterystyczny sygnał flurocentryczny.
- kolor jest rejestrowany, następnie terminator na barwniku jest rozszczepiany i myty. Proces powtarza się dla określonej liczby cykli.,
wywołanie bazowe
Illumina ma autorskie oprogramowanie, które przechodzi przez wszystkie obrazy przechwycone w poprzednim etapie i generuje pliki tekstowe z informacjami o sekwencji każdego klastra na podstawie fluorescencji. Oprócz wywoływania baz, to oprogramowanie przypisuje wynik prawdopodobieństwa, aby wskazać, jak pewne było wywołanie czegoś „A”,” T”,” G „lub”C”.
Jeśli są jakieś niejasności, np., w pewnym cyklu obraz dla klastra nie ma wyraźnego koloru, który może być związany z określonym nukleotydem, oprogramowanie wywołujące bazę będzie miało niskie prawdopodobieństwo związane z nim i przypisałoby „N” zamiast „A”, „T”, „G” lub „C”.
na zakończenie
- liczba klastrów ~= Liczba odczytów
- liczba cykli sekwencjonowania = Długość odczytów
liczba cykli (długość odczytów) zależy od użytej platformy sekwencjonowania, a także od Twoich preferencji.
Uwaga., Jeśli chcesz zgłębić sekwencjonowanie poprzez syntezę, polecamy tę naprawdę ładną animację dostępną na kanale YouTube Illumina.
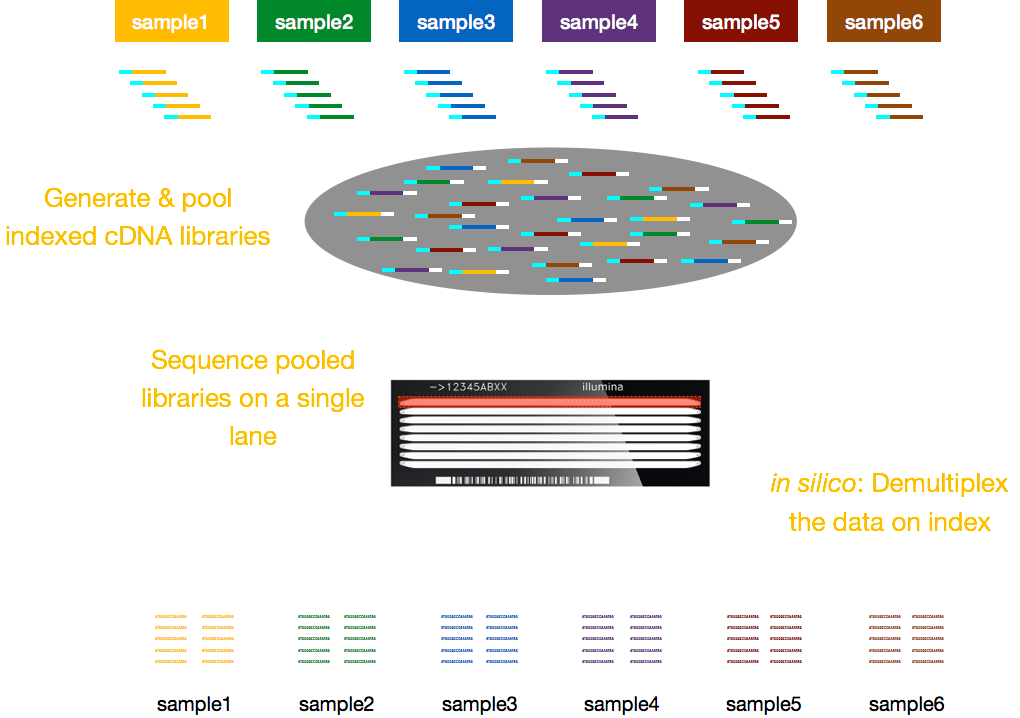
multipleksowanie
w zależności od platformy Illumina (MiSeq, HiSeq, NextSeq), liczba pasów ruchu na komórkę przepływu i liczba odczytów, które można uzyskać na pasie, różnią się znacznie. Będziesz musiał zdecydować, ile czytań chcesz na próbkę (tj. głębokość sekwencji), a następnie na podstawie wybranej platformy Oblicz, ile łącznych ścieżek będziesz potrzebować dla zestawu próbek., Porozmawiamy więcej o rozważaniach przy podejmowaniu tej decyzji w następnej lekcji na temat rozważań eksperymentalnych
zazwyczaj opłaty za sekwencjonowanie są na pasie komórki przepływowej i będziesz mógł uruchomić wiele próbek na pasie. Illumina opracowała zatem przyjemną metodę multipleksowania, która pozwala bibliotekom z kilku próbek na łączenie i sekwencjonowanie jednocześnie w tym samym pasie komórki przepływowej. Metoda ta wymaga dodania indeksów (wewnątrz adaptera Illumina) lub specjalnych kodów kreskowych (poza adapterem Illumina), jak opisano na poniższym schemacie.,
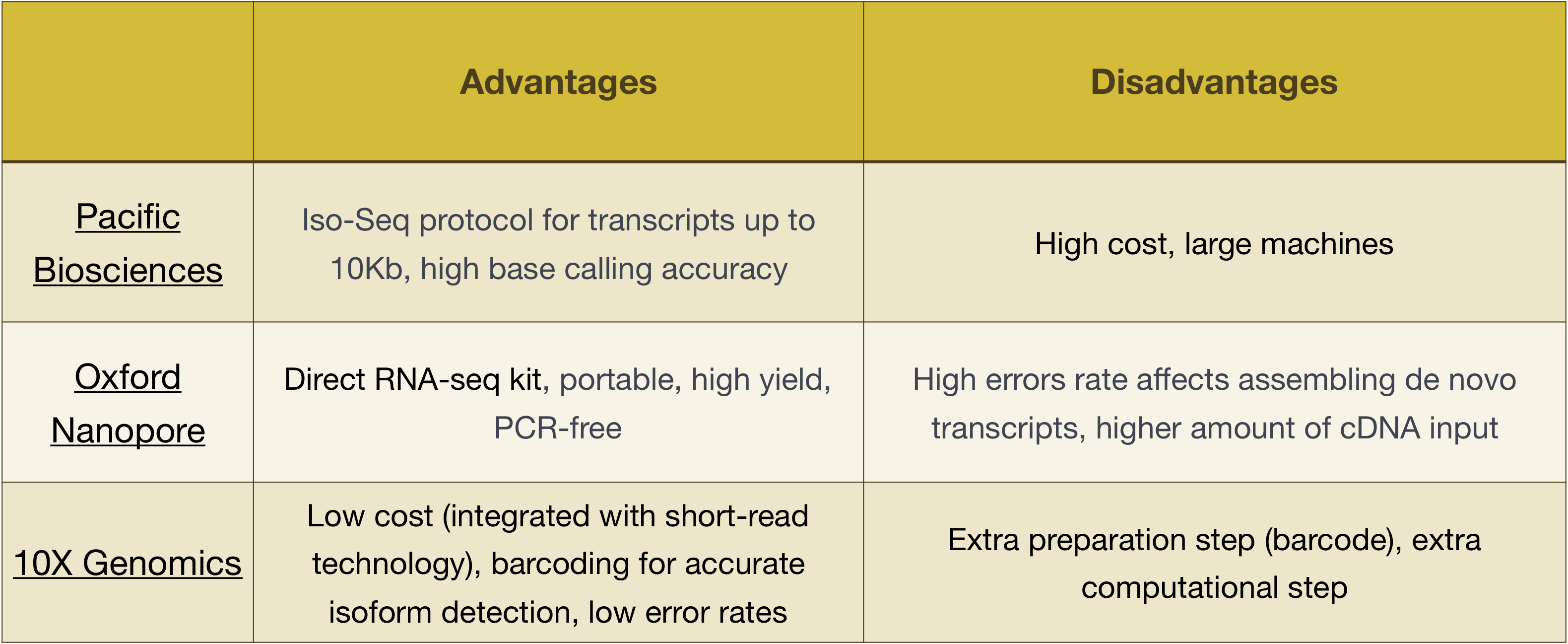
Uwaga: przepływ pracy przedstawiony w tej lekcji jest specyficzny dla sekwencjonowania Illumina, który jest obecnie najbardziej wykorzystywaną metodą sekwencjonowania., Ale są inne długo czytane metody sekwencjonowania warte odnotowania, takie jak:
- Pacific Biosciences: http://www.pacb.com/
- Nanopore Oxford (MinION): https://nanoporetech.com/
- 10x genomika: https://www.10xgenomics.com/
zalety i wady tych technologii można zbadać w tabeli poniżej:
Dodaj komentarz