hora Aproximada: 90 minutos
os Objectivos de Aprendizagem:
- Descrever o processo de RNA-seq biblioteca de preparo
- Descrever o Illumina método de seqüenciamento
Introdução à RNA-seq
RNA-seq é uma interessante técnica experimental que é utilizado para explorar e/ou quantificar a expressão do gene dentro ou entre as condições.,como sabemos, os genes fornecem instruções para fazer proteínas, que desempenham alguma função dentro da célula. Embora todas as células contenham a mesma sequência de ADN, as células musculares são diferentes das células nervosas e de outros tipos de células devido aos diferentes genes que são ligados nestas células e aos diferentes RNAs e proteínas produzidas.
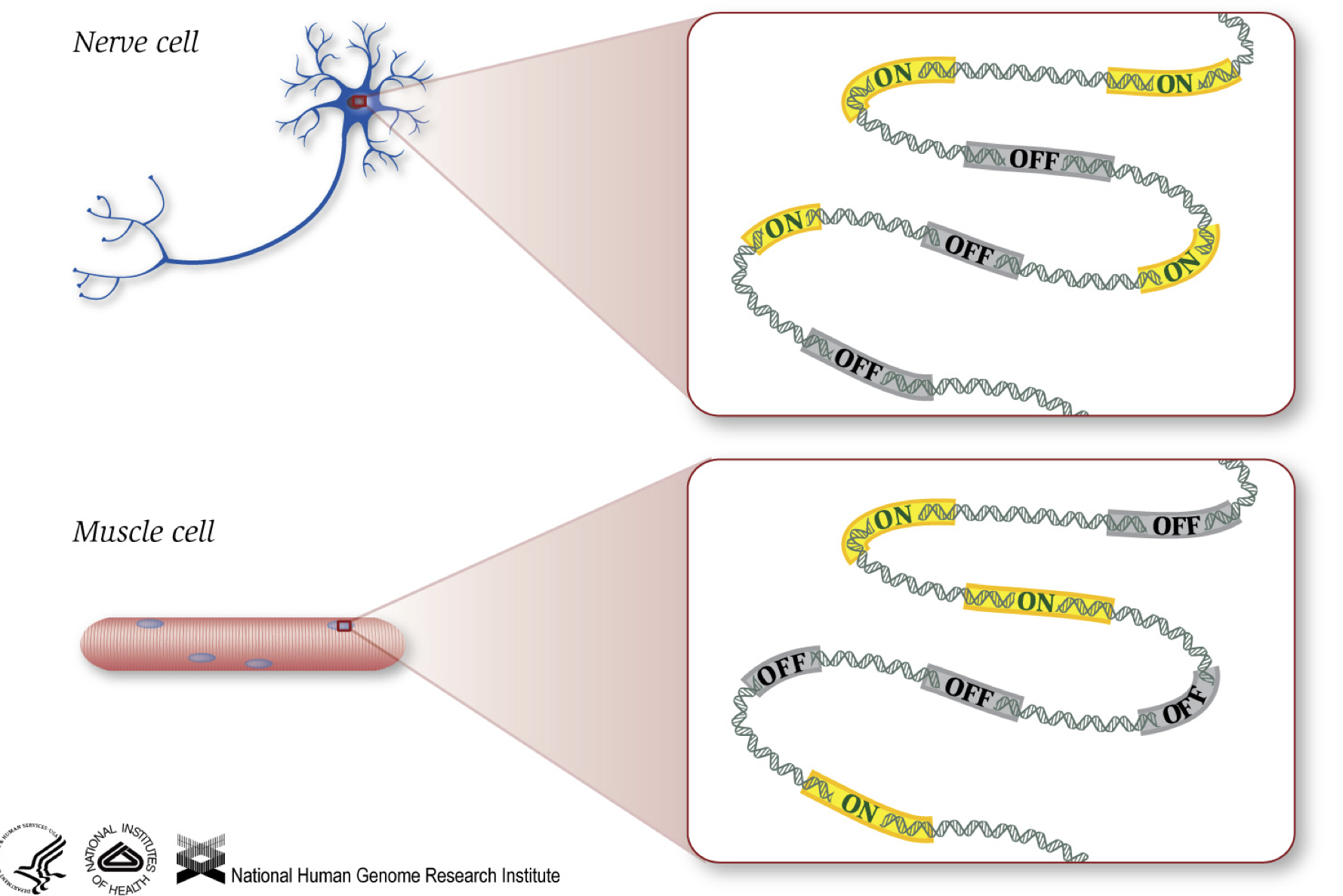
Diferentes processos biológicos, bem como mutações, podem afetar os genes que estão ligados e que estão desativados, além de, como muito genes específicos estão ligado/desligado.,
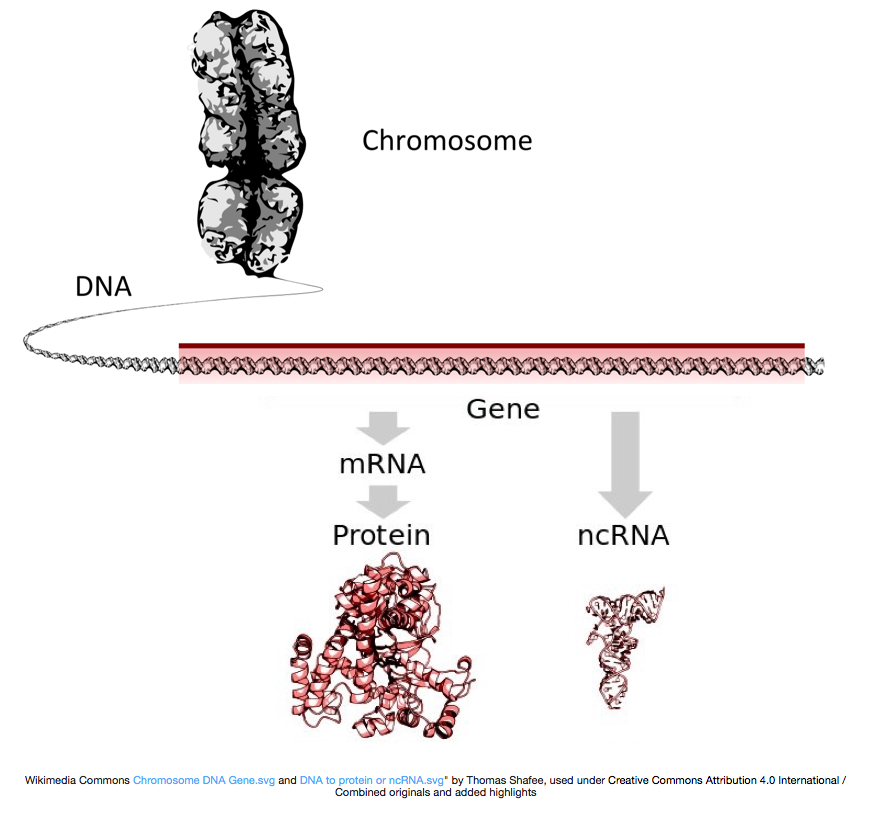
para produzir proteínas, o ADN é transcrito para ARN mensageiro, ou ARNm, que é traduzido pelo ribossoma em proteína. No entanto, alguns genes codificam o RNA que não se traduz em proteínas; estas RNAs são chamadas de RNAs não codificantes, ou ncRNAs. Muitas vezes estas RNAs têm uma função dentro e fora de si e incluem rRNAs, tRNAs, e siRNAs, entre outros. Todas as RNAs transcritas a partir de genes são chamadas transcrições.
para ser traduzido em proteínas, o ARN deve ser submetido a processamento para gerar o ARNm., Na figura abaixo, a linha superior da imagem representa um gene no DNA, composto pelas regiões não traduzidas (UTRs) e a moldura de leitura aberta. Os Genes são transcritos em pré-ARNm, que ainda contém as sequências intrónicas. Após o processamento pós-trancipcional, os intrões são spliced para fora e uma cauda de polyA e cap de 5′ são adicionados para produzir transcrições maduras de mRNA, que podem ser traduzidas em proteínas.,
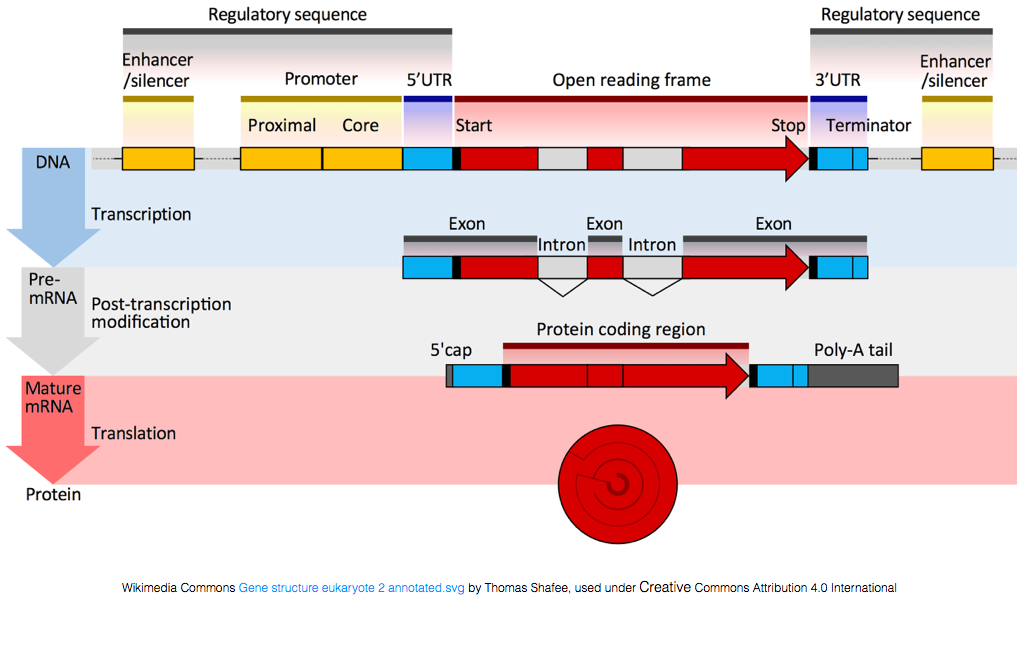
embora as transcrições de mRNA tenham uma cauda de polyA, muitas das transcrições de RNA não codificam, uma vez que o processamento pós-transcrição é diferente para estas transcrições.
Transcriptomics
a transcriptoma é definida como uma coleção de todas as leituras de transcrição presentes em uma célula., RNA-seq dados podem ser usados para explorar e/ou quantificar o transcriptoma de um organismo, que pode ser utilizado para os seguintes tipos de experimentos:
- Expressão Gênica Diferencial: quantitativos de avaliação e comparação de transcrição níveis
- Transcriptoma montagem: construção do perfil das regiões transcritas do genoma, uma avaliação qualitativa.,
- Pode ser usada para ajudar a construir a melhor gene modelos, e verificá-los usando o assembly
- Metatranscriptomics ou comunidade transcriptoma de análise
Illumina biblioteca de preparo
Ao iniciar um RNA-seq experimento, para cada amostra de RNA precisa ser isolado e se transformou em uma biblioteca de cDNA para a sequência. O fluxo de trabalho geral para a preparação da Biblioteca é detalhado nas imagens passo a passo abaixo.
brevemente, o ARN é isolado da amostra e o ADN contaminante é removido com DNase.,
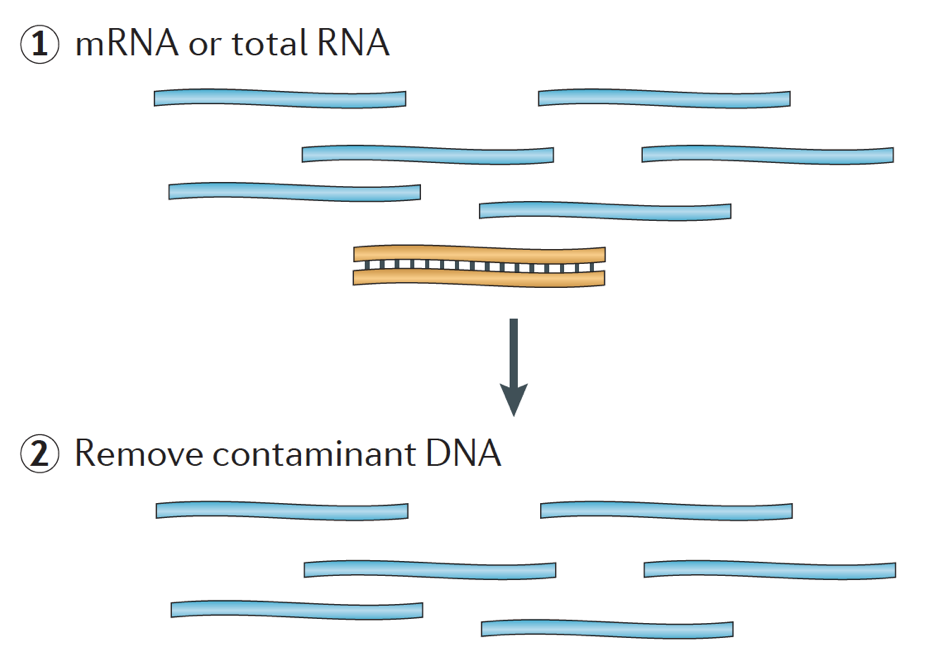
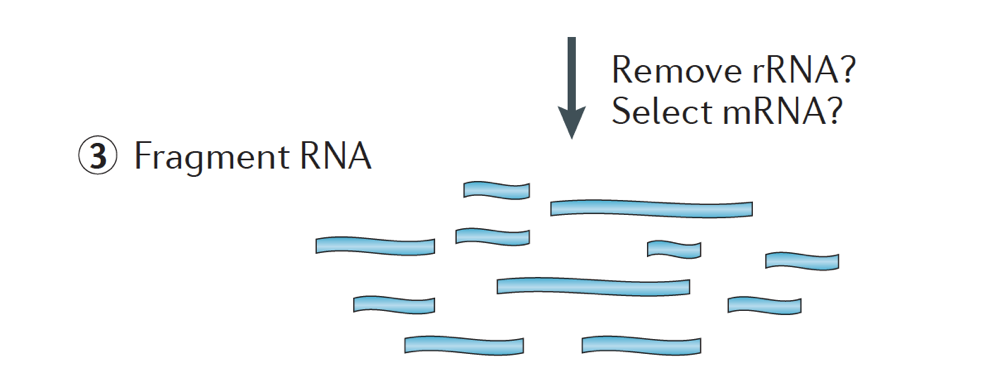
a amostra de ARN passa então pela Selecção do ARNm (selecção de polyA) ou pela depleção do ARNr. O RNA resultante é fragmentado.
geralmente, o ARN ribossómico representa a maioria das RNAs presentes numa célula, enquanto que as RNAs do Mensageiro representam uma pequena percentagem de RNA total, ~2% no ser humano. Por conseguinte, se queremos estudar os genes codificadores de proteínas, temos de enriquecer o ARNm ou esgotar o ARNr., Para a análise da expressão genética diferencial, é melhor enriquecer para o Poli (A)+, a menos que você esteja procurando obter informações sobre longas RNAs não codificantes, em seguida, fazer uma depleção ribossômica de RNA.
o tamanho dos fragmentos-alvo na biblioteca final é um parâmetro fundamental para a construção da biblioteca. A fragmentação do DNA é tipicamente feita por métodos físicos (ou seja, cisalhamento acústico e sonicação) ou por métodos enzimáticos (ou seja, coquetéis endonuclease não específicos e reações de tagmentação da transposase).,
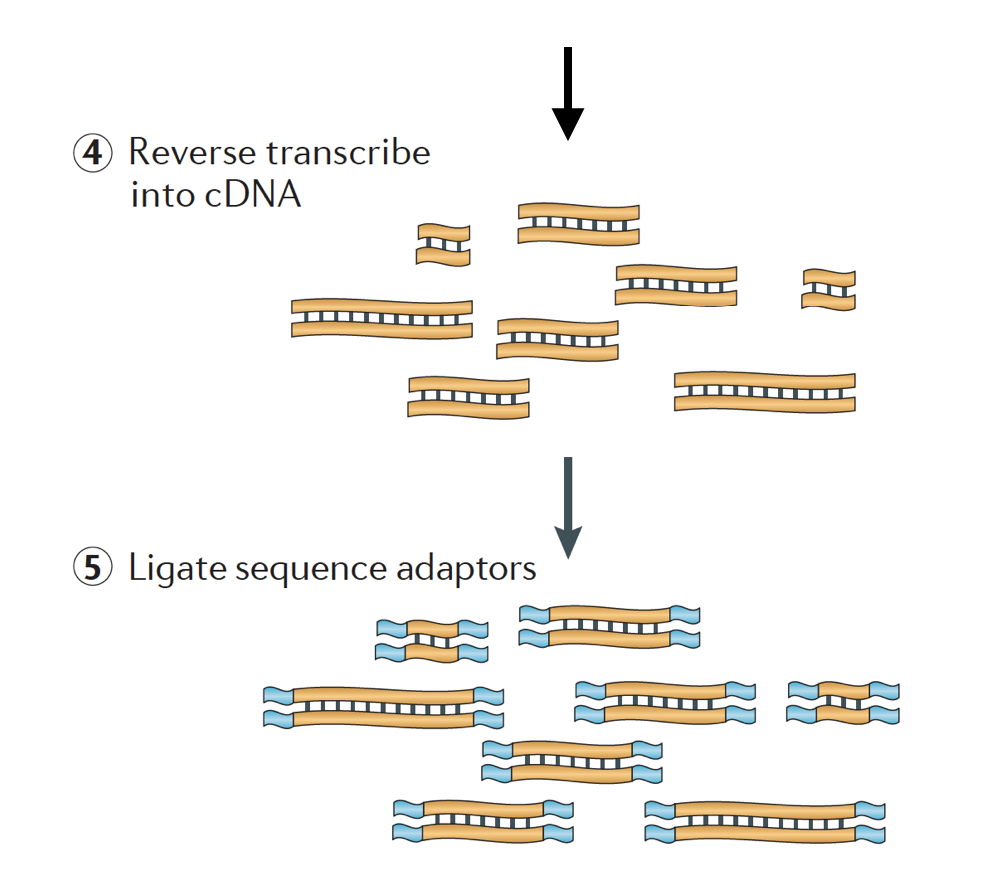
o ARN é então transcrito para trás em cDNA de cadeia dupla e Adaptadores de sequência são então adicionados às extremidades dos fragmentos.
as bibliotecas de cDNA podem ser geradas de uma forma a reter informação sobre a cadeia de ADN da qual o ARN foi transcrito. Bibliotecas que retêm essas informações são chamadas de bibliotecas encalhadas, que agora são padrão com kits TRUSEQ RNA-Seq de ilumina., As bibliotecas encalhadas não devem ser mais dispendiosas do que as que não são controladas, pelo que não há, de facto, qualquer razão para não adquirirem esta informação adicional.,
Existem 3 tipos de bibliotecas de cDNA disponível:
- para a Frente (secondstrand) – lê semelhantes à sequência de gene ou o secondstrand cDNA sequência
- Inverter (firststrand) – lê semelhantes o complemento da sequência de gene ou firststrand sequência de cDNA (TruSeq)
- Unstranded
Finalmente, os fragmentos de PCR amplificado, se necessário, e os fragmentos são de tamanho selecionado (normalmente ~300-500bp) para concluir a biblioteca.
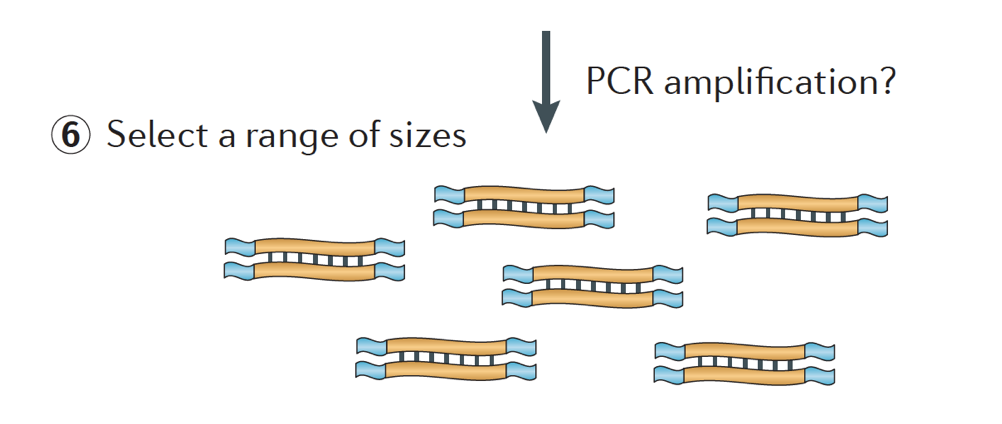
Image credit: Martin J. A. and Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina Seqüenciamento
Único-final versus Emparelhado-end
Após a preparação das bibliotecas, o seqüenciamento pode ser realizado para gerar as sequências nucleotídicas das extremidades dos fragmentos, que são chamados de leituras. Você terá a escolha de sequenciar uma única extremidade dos fragmentos cDNA (leitura de ponta única) ou ambas as extremidades dos fragmentos (leitura de ponta emparelhada).,
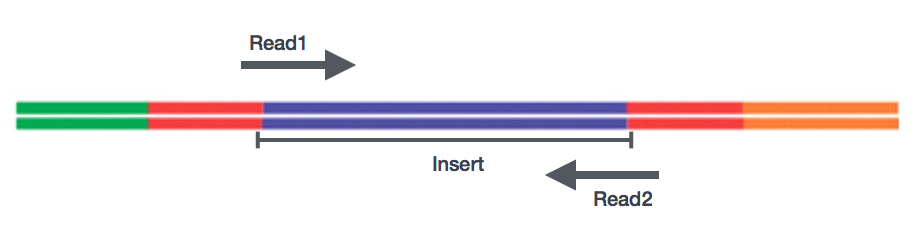
- SE – Único fim de conjunto de dados => Só Ler1
- PE – Emparelhado-end conjunto de dados => Ler1 + Read2
- pode ser de 2 em separado FastQ arquivos ou apenas uma com interleaved pares
Geralmente único fim de seqüenciamento é suficiente, a menos que o esperado é que o lê vai corresponder a vários locais no genoma (e.g. organismos com muitos paralogous genes), assembleias estão sendo realizadas, ou para emenda isoforma diferenciação. Esteja ciente de que as leituras emparelhadas são geralmente 2x mais caras.,
diferentes plataformas de sequenciamento
há uma variedade de plataformas de iluminação para escolher entre a seqüência das bibliotecas cDNA.

crédito da Imagem: Adaptado de Illumina
as Diferenças na plataforma pode alterar a duração de lê gerado, a qualidade de leituras, bem como o número total de leituras seqüenciado por execução e a quantidade de tempo necessário para a sequência de bibliotecas., As diferentes plataformas cada uma usa uma célula de fluxo diferente, que é uma superfície de vidro revestida com um arranjo de oligos emparelhados que são complementares aos adaptadores adicionados a suas moléculas modelo. A célula de fluxo é onde as reações de sequenciamento ocorrem.

crédito da Imagem: Adaptado de Illumina
Seqüenciamento-por-síntese
Illumina tecnologia de sequenciamento utiliza uma sequência-por-síntese, a abordagem que é descrita em mais detalhes abaixo.
na etapa, os fragmentos de DNA na biblioteca cDNA são desnaturados e aplicados na célula de fluxo de vidro., Estes fragmentos desnaturados ligam-se aos oligos complementares que já estão covalentemente ligados às faixas de células de fluxo, resultando em ligação.
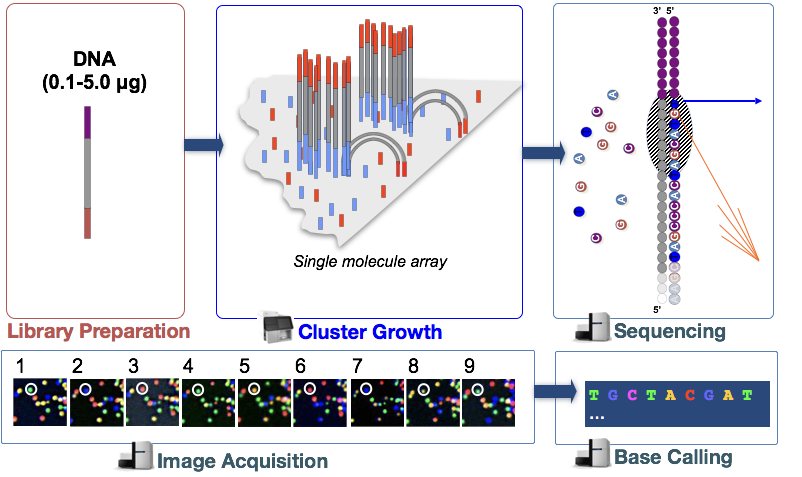
geração de clusters
Uma vez que os fragmentos tenham ligado, uma fase chamada geração de clusters começa. Durante esta etapa, fragmentos únicos são clonalmente amplificados para criar um aglomerado (fragmentos na proximidade próxima) de fragmentos idênticos. Isto é necessário para que a fluorescência possa ser facilmente capturada a partir de cada aglomerado, em vez de um único fragmento, durante a incorporação de nucleótidos na etapa seguinte.,
- sintetizar o complemento com polimerase
- repita muitas vezes para amplificar clonalmente todos os fragmentos únicos na célula de fluxo para formar aglomerados de sequência idêntica.,
dsDNA é desnaturado e o ADN original é lavado, deixando a cadeia sintetizada ligada covalentemente à célula de fluxo.a cadeia única hibridiza com adaptador adjacente para formar uma ponte dsDNA é estendida por polimerase. Cada cadeia ligada covalentemente a um adaptador diferente.
Sequenciamento por síntese (& aquisição de imagem)
Depois de cluster de geração de microesferas marcados de nucleotídeos são incorporados a um de cada vez (ciclicamente) e de fluorescência imagens são capturadas para identificar o que de nucleotídeos fica incorporado em cada cluster em cada ciclo.
- aglomerados de desnaturação e o bloco 3′ termina para evitar a preparação indesejada.iniciadores de sequenciação de hibridação para sequência de adaptadores nas pontas soltas.,ciclo quatro NTPs com marcadores fluorescentes e sequência terminadora e polimerases.uma vez que NTP é incorporado, o aglomerado é animado por uma fonte de luz e um sinal fluroscente característico é emitido.
- A cor é gravada, em seguida, o terminador no corante é clivado e lavado. Repete-se o processo para o número específico de ciclos.,
Base Calling
Illumina tem software proprietário que passa por todas as imagens capturadas na fase anterior e gera arquivos de texto com informações de sequência sobre cada aglomerado com base na fluorescência. Além de chamar as bases, este software atribui uma pontuação probablity para indicar o quão certo era sobre a chamada de algo um “A”, um “T”, Um “G” ou um “C”.
Se houver ambiguidades, por exemplo:, em um determinado ciclo, a imagem de um cluster não tem uma cor distinta que pode ser associada com um nucleótido específico, o software de chamada de base terá uma baixa probabilidade associada a ele e atribuiria um “N” em vez de “A”, “T”, “G” ou “C”.
No fechamento
- Número de clusters ~= Número de leituras
- Número de sequenciamento de ciclos = Comprimento de lê
O número de ciclos (comprimento das leituras) dependerá de seqüenciamento plataforma utilizada, bem como as suas preferências.
nota., Se você quiser explorar a sequenciação por síntese em mais profundidade, recomendamos esta animação realmente agradável disponível no canal da Illumina’s YouTube.
Multiplexação
Dependendo da plataforma Illumina (MiSeq, HiSeq, NextSeq), o número de faixas por célula de fluxo, e o número de leituras que podem ser obtidos por via varia amplamente. Você terá que decidir quantas leituras você gostaria por amostra (ou seja, a profundidade de sequenciamento) e, em seguida, com base na plataforma que você escolher calcular quantas faixas totais você vai precisar para o seu conjunto de amostras., Nós falaremos mais sobre considerações ao tomar esta decisão na próxima lição sobre considerações experimentais
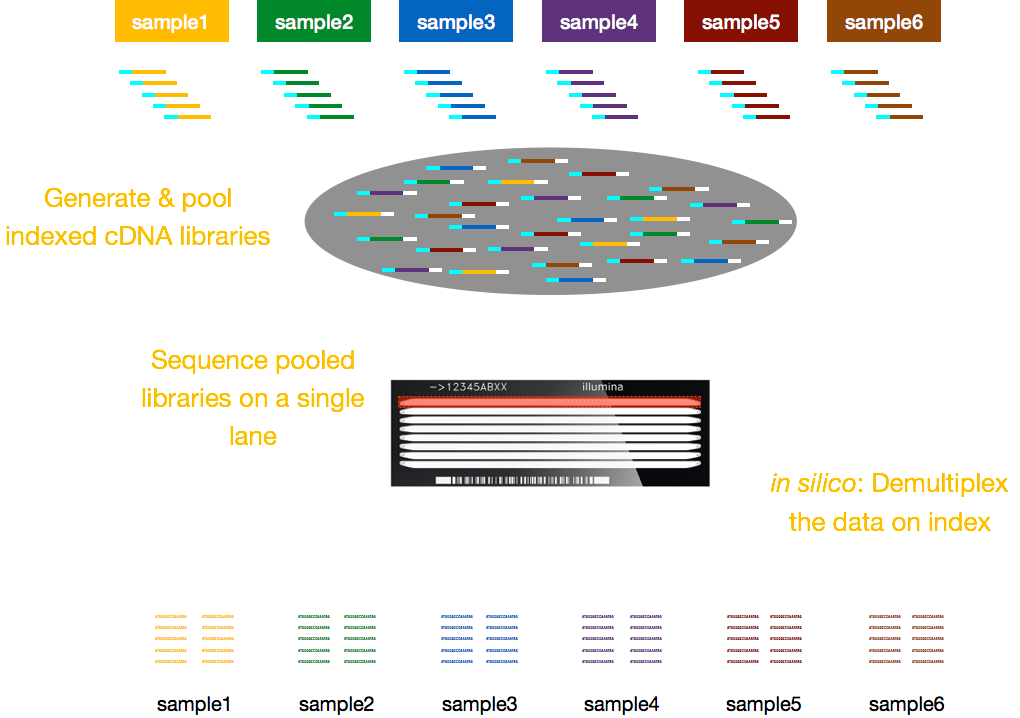
tipicamente, cargas para sequenciamento são por faixa da célula de fluxo e você será capaz de executar várias amostras por faixa. Illumina desenvolveu, portanto, um bom método de multiplexagem que permite que bibliotecas de várias amostras sejam agrupadas e sequenciadas simultaneamente na mesma faixa de uma célula de fluxo. Este método requer a adição de índices (dentro do adaptador Illumina) ou códigos de barras especiais (fora do adaptador Illumina), conforme descrito no esquema abaixo.,
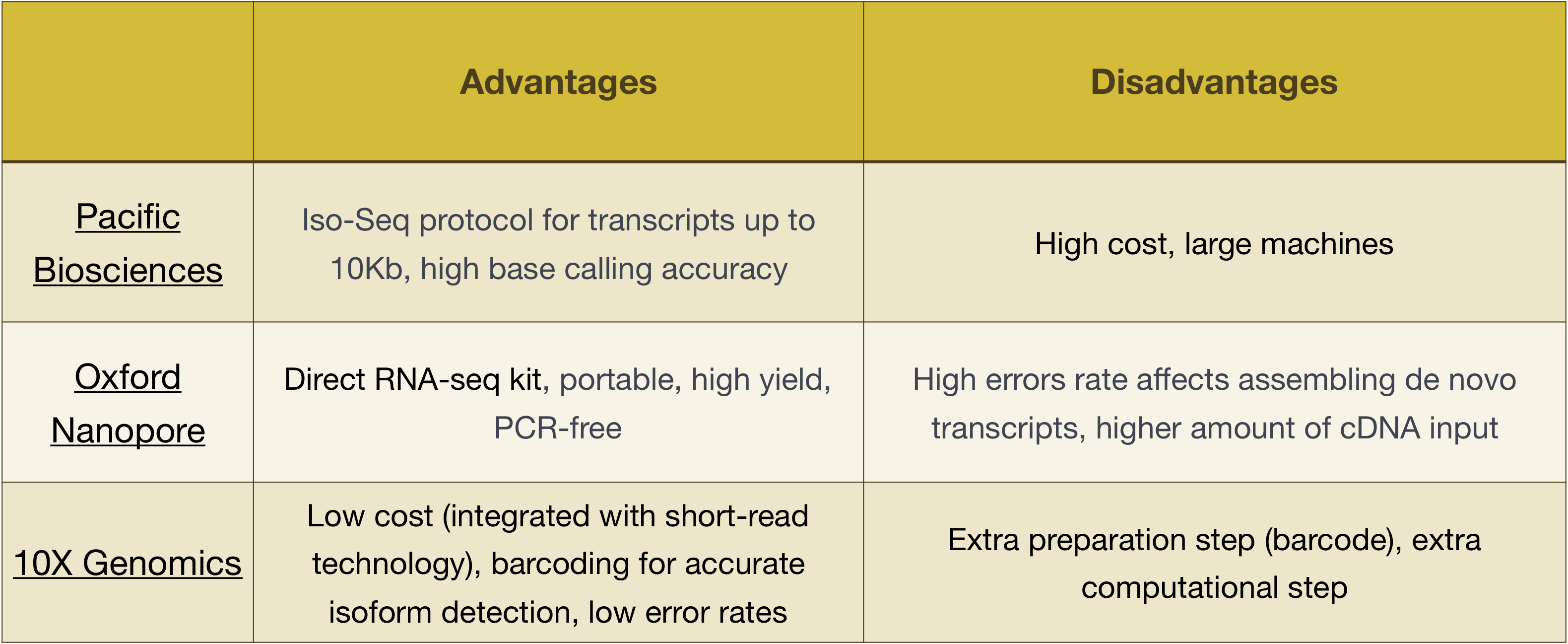
NOTA: O fluxo de trabalho apresentados nesta lição é específico para o Illumina de seqüenciamento, que é atualmente o mais utilizado método de seqüenciamento., Mas há outros longa-leitura de sequenciamento de métodos digno de nota, tais como:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genômica: https://www.10xgenomics.com/
Vantagens e desvantagens destas tecnologias pode ser explorado na tabela abaixo:
Deixe uma resposta