ungefärlig tid: 90 minuter
inlärningsmål:
- beskriv processen med RNA-seq-bibliotekspreparat
- beskriv Illumina-sekvenseringsmetoden
introduktion till RNA-seq
RNA-seq är en spännande och spännande introduktion till RNA-seq.experimentell teknik som används för att utforska och/eller kvantifiera genuttryck inom eller mellan betingelser.,
som vi vet ger gener instruktioner för att göra proteiner, som utför någon funktion inom cellen. Även om alla celler innehåller samma DNA-sekvens, muskelceller skiljer sig från nervceller och andra typer av celler på grund av de olika gener som slås på i dessa celler och de olika RNA och proteiner som produceras.
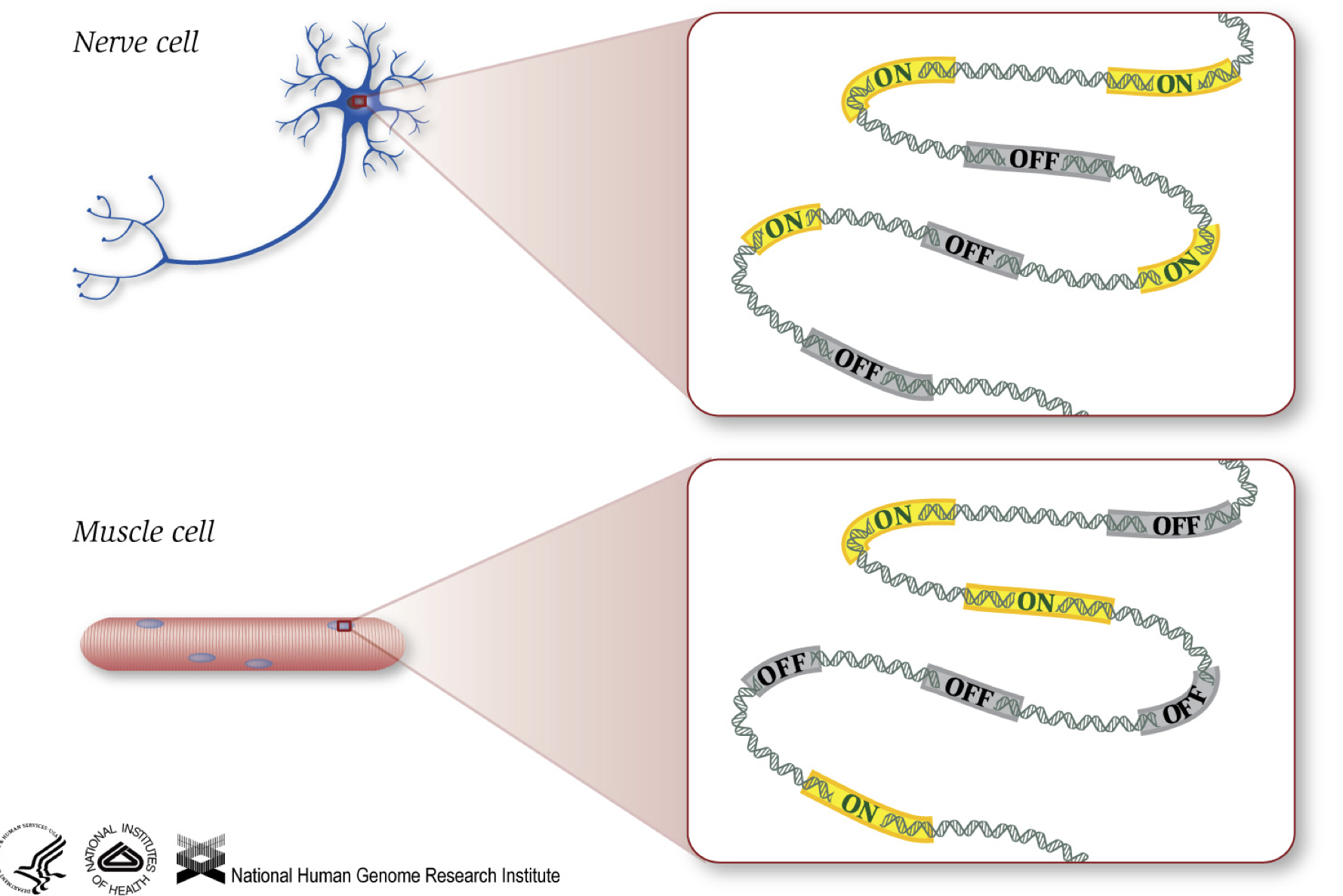
olika biologiska processer, såväl som mutationer, kan påverka vilka gener som är påslagna och som är avstängda, förutom hur mycket specifika gener som är på / av.,
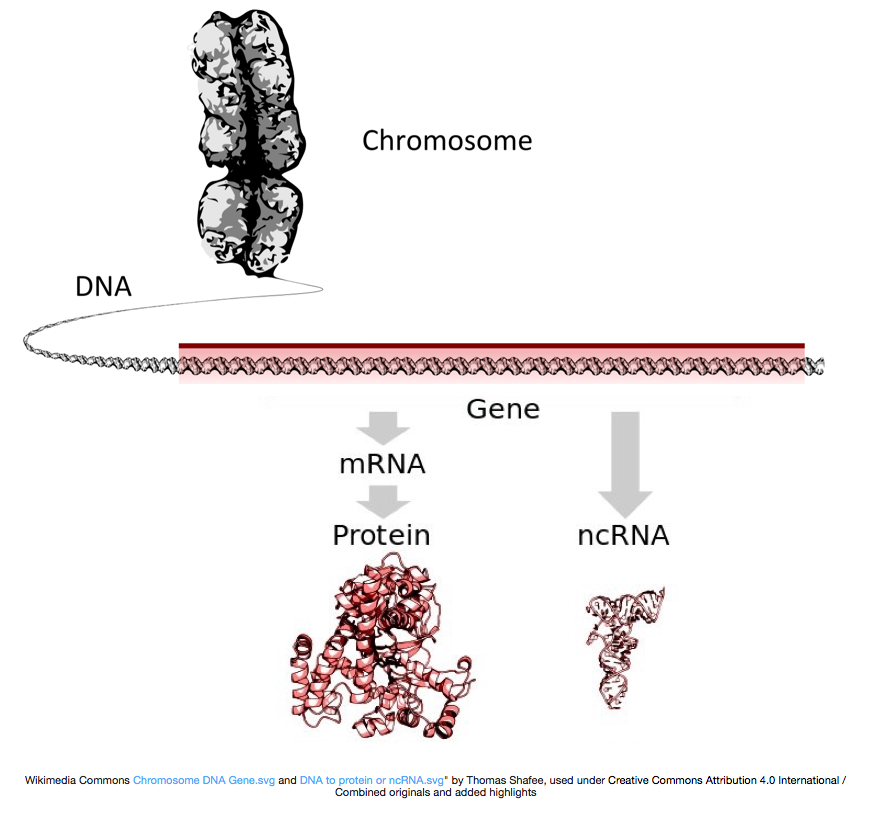
för att göra proteiner transkriberas DNA till messenger RNA, eller mRNA, som översätts av ribosomen till protein. Men vissa gener kodar RNA som inte blir översatt till protein; dessa rna kallas icke-kodande RNA, eller ncRNAs. Ofta har dessa RNAs en funktion i och av sig själva och inkluderar rRNAs, tRNAs och siRNAs, bland andra. Alla RNA transkriberas från gener kallas transkript.
för att översättas till proteiner måste RNA genomgå bearbetning för att generera mRNA., I figuren nedan representerar den övre strängen i bilden en gen i DNA, bestående av de otranslaterade regionerna (UTRs) och den öppna läsramen. Gener transkriberas till pre-mRNA, som fortfarande innehåller de introniska sekvenserna. Efter transciptional bearbetning splitsas intronerna ut och en polyA svans och 5′ keps läggs till för att ge mogna mRNA-transkript, som kan översättas till proteiner.,
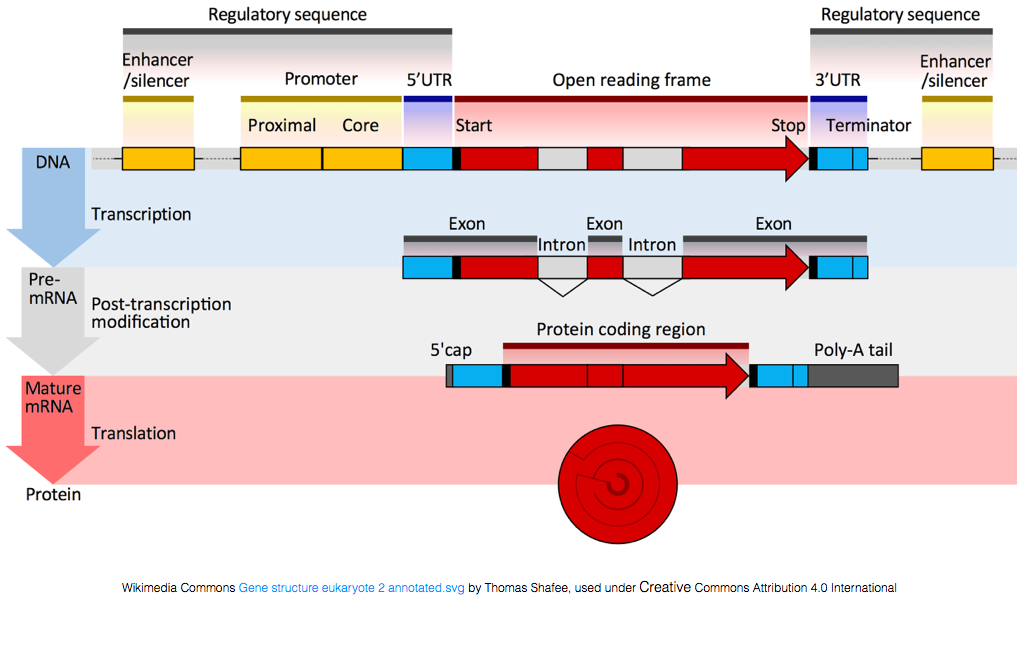
medan mRNA-transkript har en polyA svans, gör många av de icke-kodande RNA-transkripten inte eftersom den post-transkriptionella behandlingen är annorlunda för dessa transkript.
Transcriptomics
transcriptome definieras som en samling av alla transkriptavläsningar som finns i en cell., RNA-seq data kan användas för att utforska och/eller kvantifiera transkriptom av en organism, som kan användas för följande typer av experiment:
- Differential genuttryck: kvantitativ utvärdering och jämförelse av transkriptionsnivåer
- Transkriptommontering: bygga profilen av transkriberade regioner av genomet, en kvalitativ utvärdering.,
- kan användas för att bygga bättre genmodeller och verifiera dem med hjälp av enheten
- Metatranscriptomics eller community transcriptome analysis
Illumina library preparation
När man startar ett RNA-seq-experiment måste RNA för varje prov isoleras och omvandlas till ett cDNA-bibliotek för sekvensering. Det allmänna arbetsflödet för biblioteksförberedelse beskrivs i steg-för-steg-bilderna nedan.
kort sagt isoleras RNA från provet och kontaminerande DNA avlägsnas med DNas.,
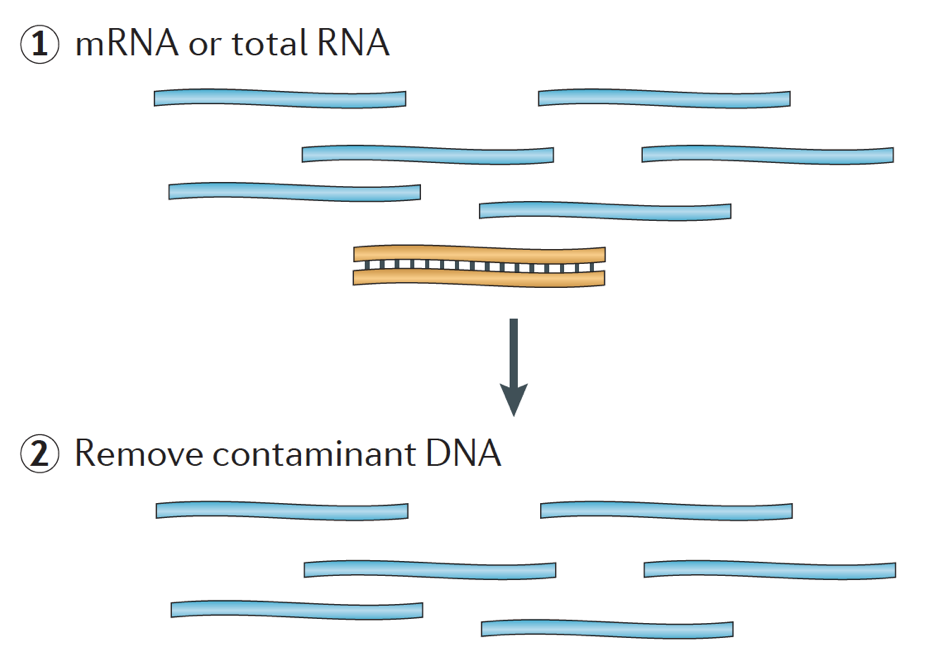
RNA-provet genomgår sedan antingen val av mRNA (polyA selection) eller utarmning av rRNA. Det resulterande RNA är fragmenterat.
generellt representerar ribosomal RNA majoriteten av RNA som finns i en cell, medan messenger RNA representerar en liten andel av total RNA, ~2% hos människor. Därför, om vi vill studera proteinkodningsgener, måste vi berika för mRNA eller tömma rRNA., För differentiell genuttrycksanalys är det bäst att berika för Poly(A)+, om du inte syftar till att få information om långa icke-kodande RNA, gör sedan en ribosomal RNA-utarmning.
storleken på målfragmenten i det slutliga biblioteket är en nyckelparameter för bibliotekets konstruktion. DNA-fragmentering sker vanligen genom fysiska metoder (dvs, akustisk klippning och ultraljudsbehandling) eller enzymatiska metoder (dvs, icke-specifik endonuclease cocktails och transposase tagmentation reaktioner.,
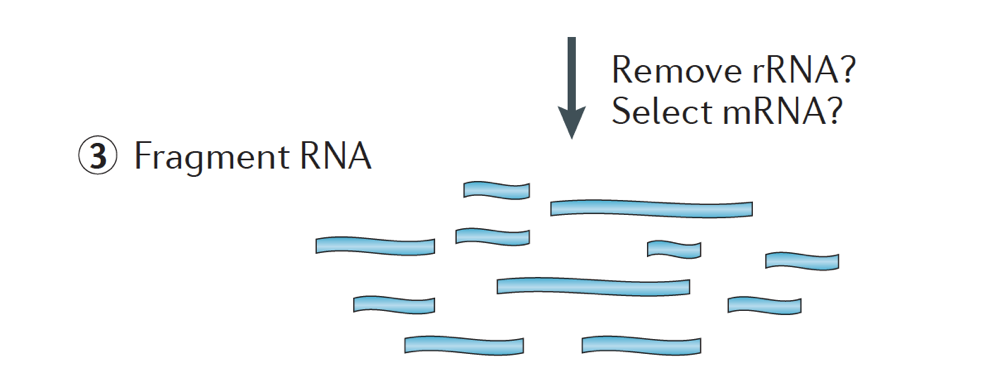
RNA är sedan omvänd transkriberas till dubbelsträngade cDNA och sekvensadaptrar läggs sedan till ändarna av fragmenten.
cDNA-biblioteken kan genereras på ett sätt att behålla information om vilken del av DNA RNA som transkriberades från. Bibliotek som behåller denna information kallas stranded libraries, som nu är standard med Illuminas TruSeq stranded RNA-Seq Kit., Stranded bibliotek bör inte vara dyrare än ostadig, så det finns egentligen ingen anledning att inte förvärva denna ytterligare information.,
det finns 3 typer av cDNA – bibliotek tillgängliga:
- framåt (secondstrand) – läser liknar gensekvensen eller secondstrand cDNA-sekvensen
- omvänd (firststrand) – läser liknar komplementet av gensekvensen eller firststrand cDNA-sekvensen (TruSeq)
- ostadig
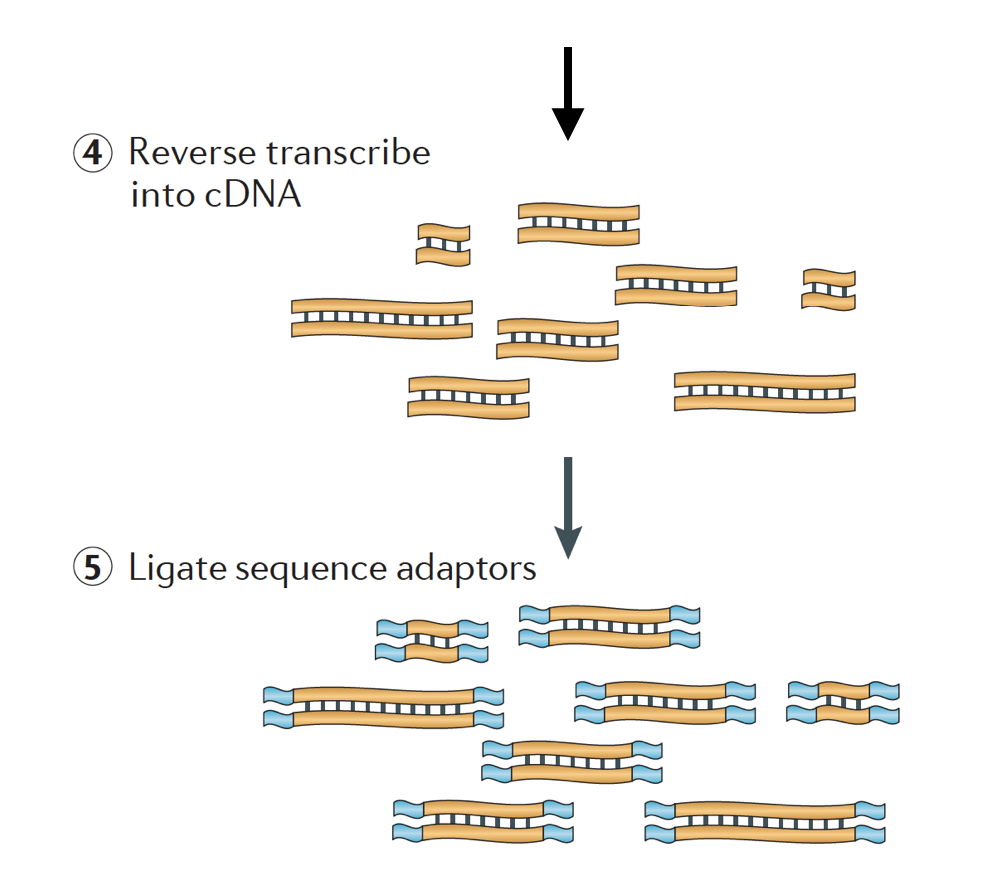
slutligen förstärks fragmenten om det behövs, och fragmenten är storlek valda (vanligtvis ~300-500bp) för att avsluta biblioteket.
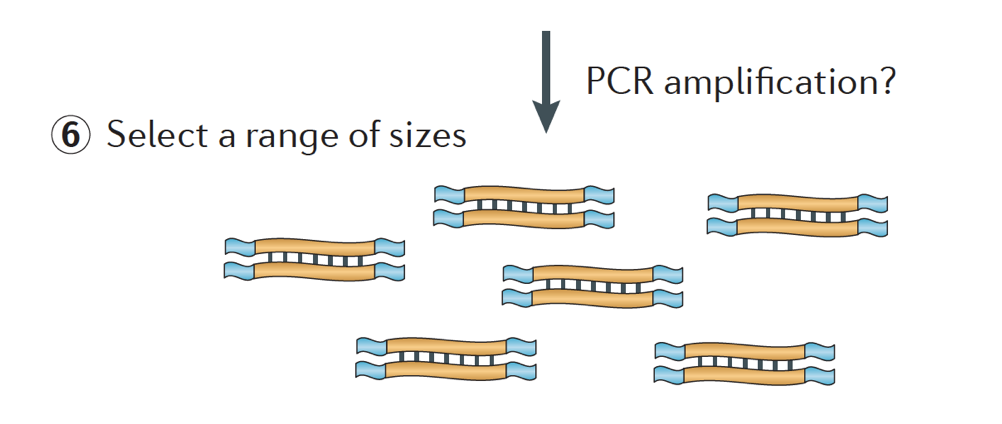
bildkredit: Martin J. A. och Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina sekvensering
Single-end versus Paired-end
efter beredning av biblioteken kan sekvensering utföras för att generera nukleotidsekvenserna av ändarna av fragmenten, som kallas läser. Du kommer att ha valet av sekvensering av en enda ände av cDNA-fragmenten (enstaka läsning) eller båda ändarna av fragmenten (Parade slutläsningar).,
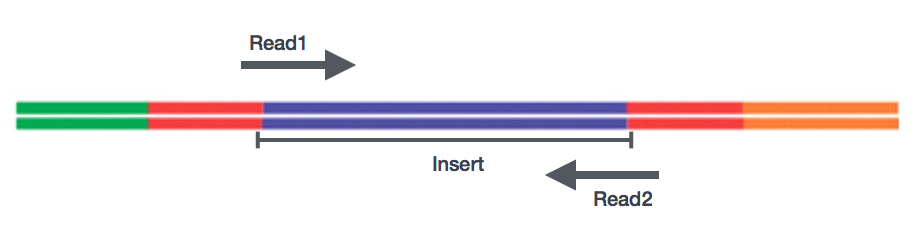
- se – Single end dataset => endast Read1
- PE – Paired-end dataset => Read1 + Read2
- kan vara 2 separata FastQ-filer eller bara en med interfolierade par
generellt är en-end-sekvensering tillräcklig om det inte förväntas att läsarna matchar flera platser på genomet (t.ex. organismer med många paralogiska gener), aggregat utförs eller för skarvsisoformdifferentiering. Var medveten om att Parade slutläsningar i allmänhet är 2x dyrare.,
olika sekvenseringsplattformar
det finns en mängd Illumina-plattformar att välja mellan för att sekvensera cDNA-biblioteken.

bildkredit: anpassad från Illumina
skillnader i plattform kan ändra längden på genererade läsningar, kvaliteten på läsningar, samt det totala antalet läsningar sekvenserade per körning och den tid som krävs för att sekvensera biblioteken., De olika plattformarna använder var och en en annan flödescell, som är en glasyta belagd med ett arrangemang av parade oligos som kompletterar de adaptrar som läggs till dina mallmolekyler. Flödescellen är där sekvenseringsreaktionerna äger rum.

bildkredit: anpassad från Illumina
sekvensering-för-syntes
Illumina-sekvenseringsteknik använder en sekvensering-för-syntes-metod som beskrivs mer detaljerat nedan.
i steget denatureras DNA-fragmenten i cDNA-biblioteket och appliceras på glasflödescellen., Dessa denaturerade fragment binder till de kompletterande oligoerna som redan är kovalent bundna till flödescellbanorna, vilket resulterar i fastsättning.
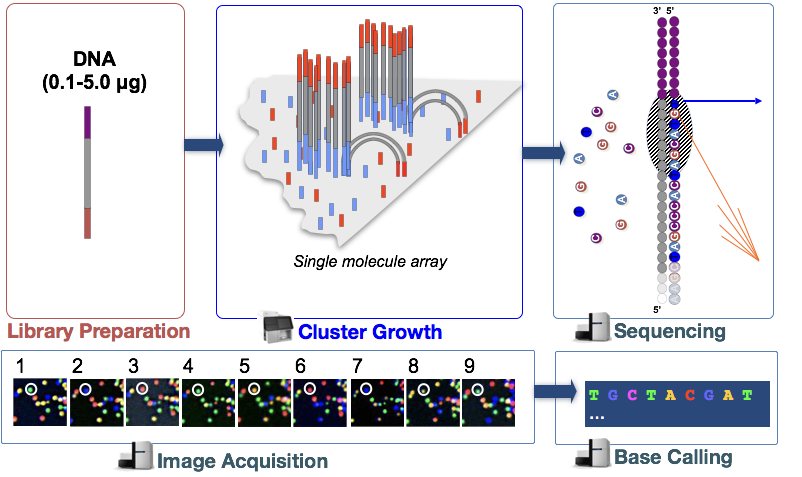
Klustergenerering
när fragmenten har bifogats börjar en fas som kallas klustergenerering. Under detta steg förstärks enskilda fragment klonalt för att skapa ett kluster (fragment i närheten) av identiska fragment. Detta är nödvändigt så att fluorescensen lätt kan fångas från varje kluster, i stället för ett enda fragment, under nukleotid införlivande i nästa steg.,
- syntetisera komplementet med polymeras
- dsDNA denatureras, och ursprungliga DNA tvättas bort lämnar syntetiserade sträng kovalent bundet till flödescell.
- single strand hybridises med intilliggande adapter för att bilda en ” bro ”
- dsDNA förlängs med polymeras. Varje sträng kovalent bunden till olika adapter.
- upprepa många gånger för att klonalt förstärka alla unika fragment på flödescellen för att bilda kluster av identisk sekvens.,
sekvensering genom syntes (& bildförvärv)
efter klustergenerering införlivas fluorescentmärkta nukleotider en i taget (cykliskt) och fluorescensbilder fångas för att identifiera vilka nukleotider som införlivas i varje kluster i varje cykel.
- Denature kluster och blocket 3′ slutar för att förhindra oönskade priming.
- hybridisera sekvensering primers till adapter sekvens vid de lösa ändarna.,
- cykel fyra NTPs med fluorescerande markörer och terminator sekvens och polymeraser.
- när NTP är införlivat, är klustret upphetsat av en ljuskälla och en karakteristisk fluroscent signal avges.
- färgen registreras, sedan terminatorn på färgämnet klyvs och tvättas. Process upprepas för specifioed antal cykler.,
Base Calling
Illumina har proprietär programvara som går igenom alla bilder som tagits i föregående steg och genererar textfiler med sekvensinformation om varje kluster baserat på fluorescensen. Förutom att ringa baserna, tilldelar denna programvara en probablity poäng för att ange hur säker det handlade om att kalla något en ”A”, En ”t”, en ”G”eller en ”C”.
om det finns några tvetydigheter, t. ex., vid en viss cykel bilden för ett kluster inte har en distinkt färg som kan associeras med en specifik nukleotid, bas ringer programvara kommer att ha en låg sannolikhet i samband med det och skulle tilldela en ”N” i stället för ”A”, ”T”, ”G” eller ”C”.
i stängning,
- antal kluster ~= antal läsningar
- antal sekvenseringscykler = längden på läsningar
antalet cykler (längden på läsningen) beror på sekvenseringsplattform som används samt dina önskemål.
OBS!, Om du vill utforska sekvensering genom syntes i mer djup, rekommenderar vi denna riktigt trevlig animation tillgänglig på Illumina YouTube-kanal.
multiplexering
beroende på Illumina-plattformen (MiSeq, HiSeq, NextSeq), antalet körfält per flödescell och antalet läsningar som kan erhållas per körfält varierar kraftigt. Du måste bestämma hur många läser du vill ha per prov (dvs sekvenseringsdjupet) och sedan baserat på plattformen väljer du beräkna hur många totala körfält du behöver för din uppsättning prover., Vi kommer att prata mer om överväganden när du fattar detta beslut i nästa lektion om experimentella överväganden
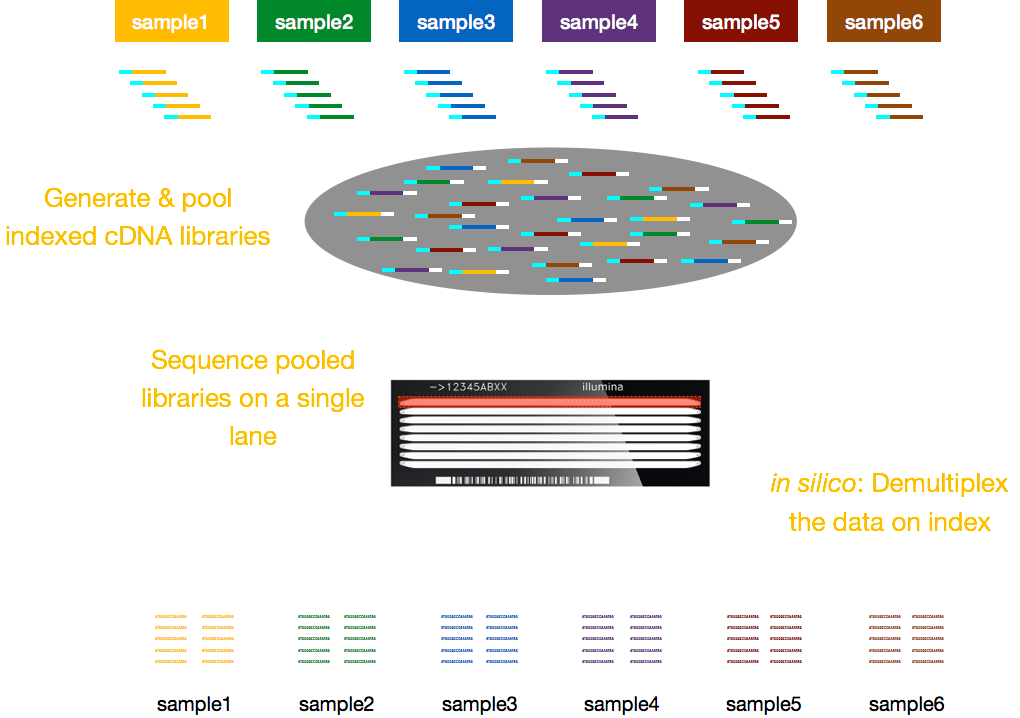
vanligtvis är avgifter för sekvensering per körfält i flödescellen och du kommer att kunna köra flera prov per körfält. Illumina har därför utarbetat en fin multiplexeringsmetod som gör att bibliotek från flera prover kan Poolas och sekvenseras samtidigt i samma körfält i en flödescell. Denna metod kräver tillsats av index (inom Illumina-adaptern) eller speciella streckkoder (utanför Illumina-adaptern) enligt beskrivningen i schemat nedan.,
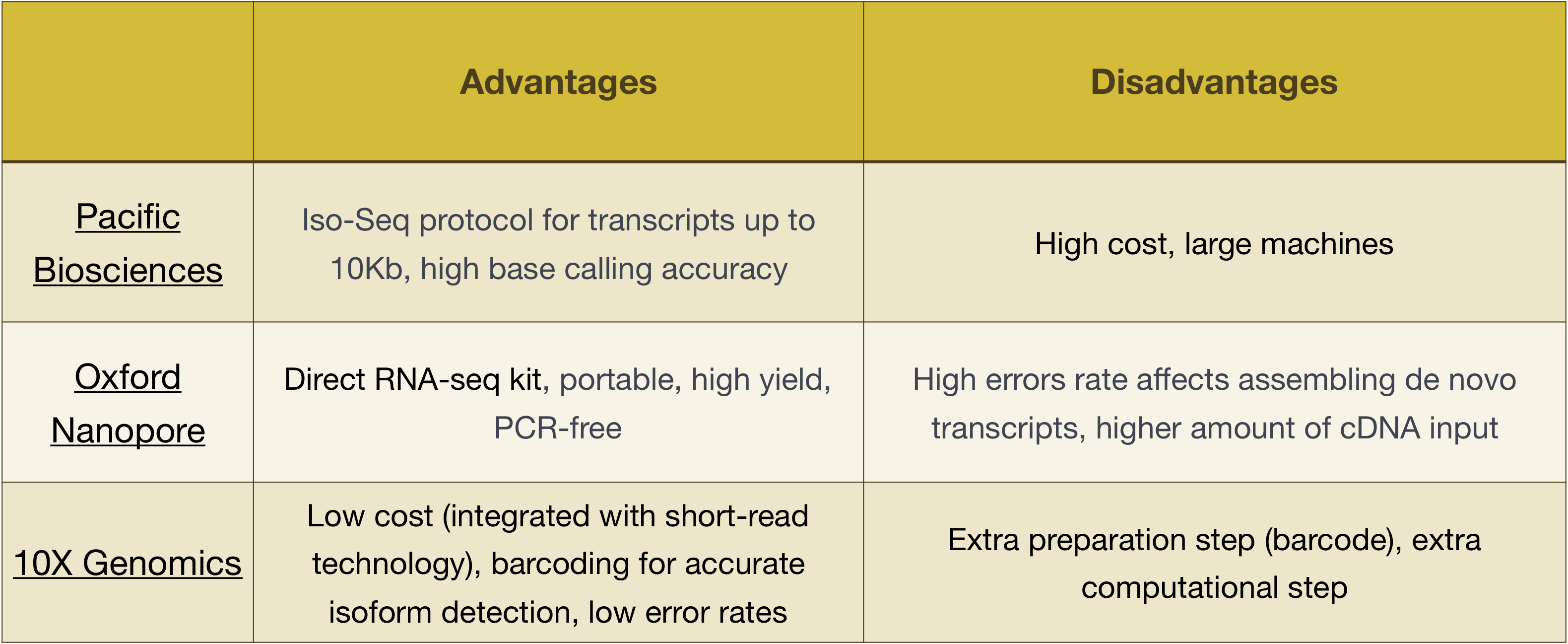
OBS! arbetsflödet som presenteras i den här lektionen är specifikt för Illumina-sekvensering, vilket för närvarande är den mest använda sekvenseringsmetoden., Men det finns andra långlästa sekvenseringsmetoder som är värda att notera, till exempel:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomics: https://www.10xgenomics.com/
fördelar och nackdelar med dessa teknik kan utforskas i tabellen nedan:
Lämna ett svar