Ungefähre Zeit: 90 Minuten
Lernziele:
- Beschreiben Sie den Prozess der RNA-seq library preparation
- Beschreiben Sie die Illumina-Sequenzierung Methode
Einführung in die RNA-seq
RNA-seq ist eine spannende experimentelle Technik, die verwertet wird, um zu erkunden und/oder Quantifizierung der Genexpression innerhalb oder zwischen den Bedingungen.,
Wie wir wissen, geben Gene Anweisungen zur Herstellung von Proteinen, die innerhalb der Zelle eine Funktion erfüllen. Obwohl alle Zellen die gleiche DNA-Sequenz enthalten, unterscheiden sich Muskelzellen von Nervenzellen und anderen Zelltypen aufgrund der verschiedenen Gene, die in diesen Zellen aktiviert sind, und der verschiedenen produzierten RNAs und Proteine.
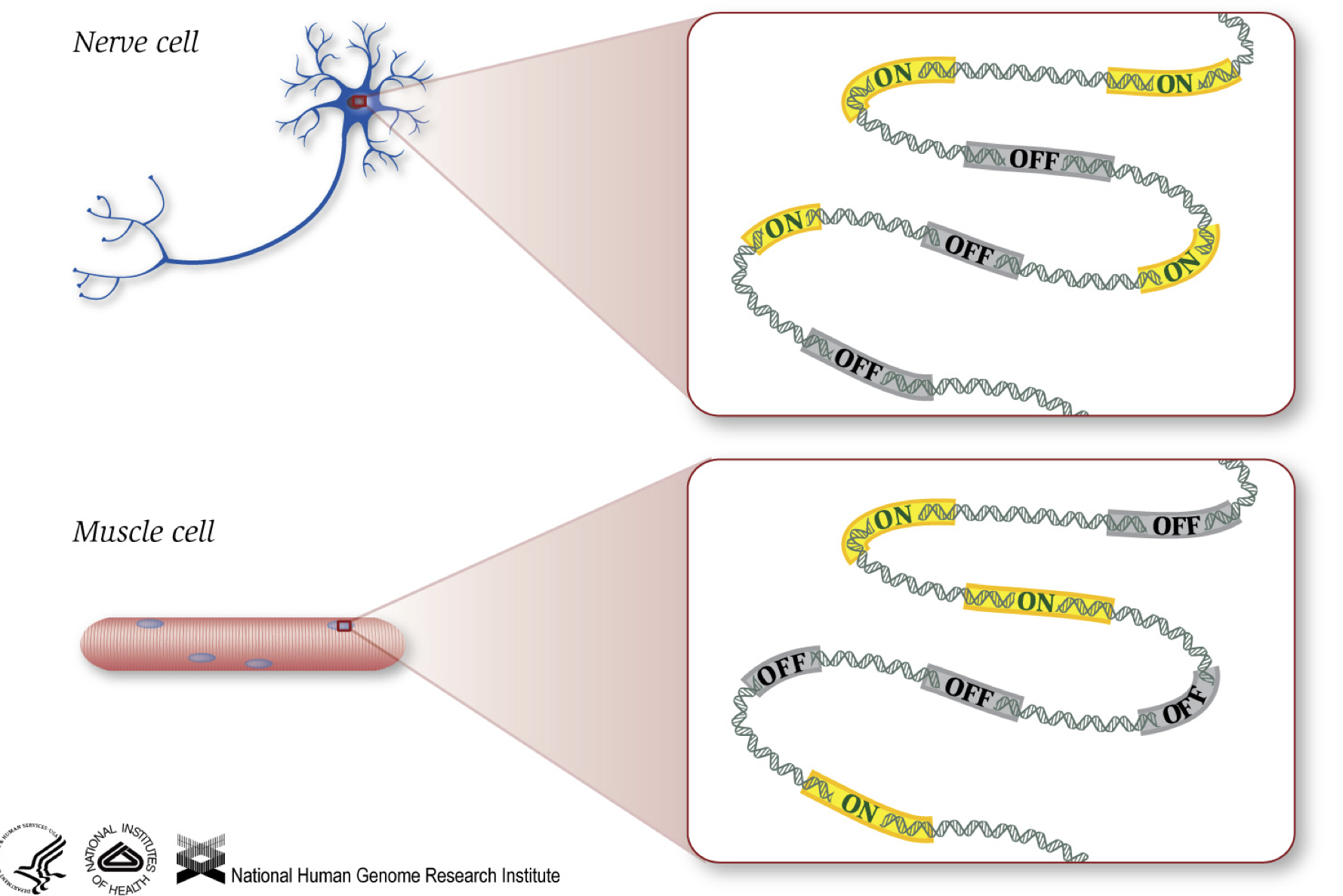
Verschiedene biologische Prozesse sowie Mutationen können beeinflussen, welche Gene eingeschaltet und welche ausgeschaltet sind, zusätzlich zu, wie viel spezifische Gene ein – /ausgeschaltet sind.,
Um Proteine herzustellen, wird die DNA in Messenger-RNA oder mRNA transkribiert, die vom Ribosom in Protein übersetzt wird. Einige Gene kodieren jedoch für RNA, die nicht in Protein übersetzt wird; Diese RNAs werden als nicht kodierende RNAs oder ncRNAs bezeichnet. Oft haben diese RNAs eine Funktion an und für sich und umfassen unter anderem rRNAs, tRNAs und siRNAs. Alle von Genen transkribierten RNAs werden Transkripte genannt.
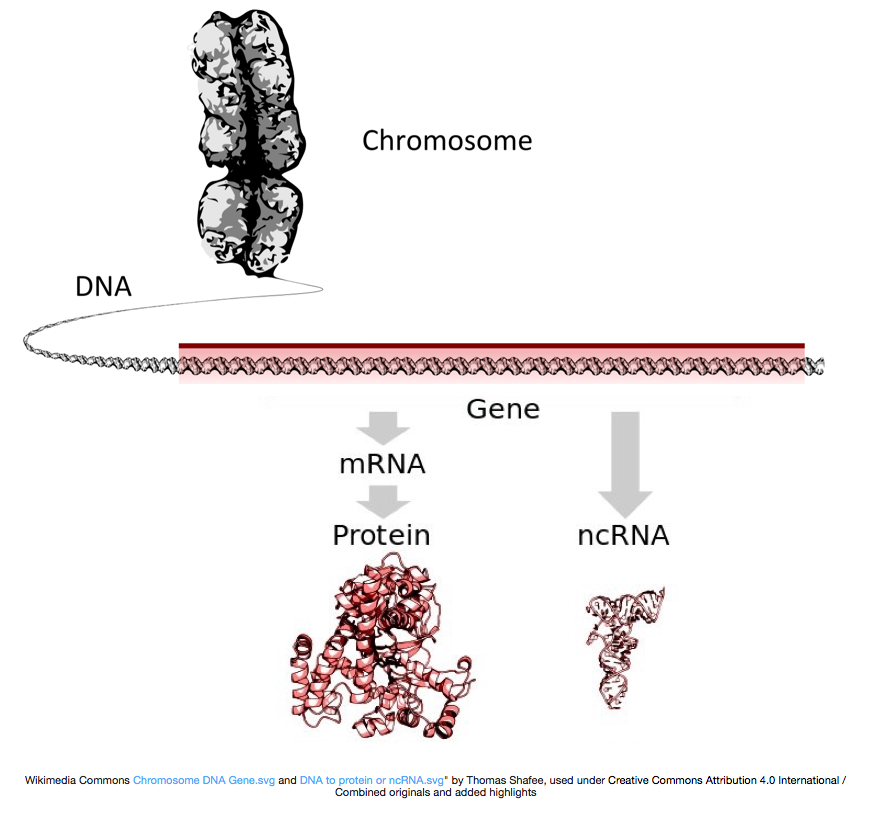
Um in Proteine übersetzt zu werden, muss die RNA zur Erzeugung der mRNA verarbeitet werden., In der folgenden Abbildung stellt der obere Strang im Bild ein Gen in der DNA dar, das aus den nicht übersetzten Regionen (UTRs) und dem offenen Leserahmen besteht. Gene werden in Prä-mRNA transkribiert, die noch die intronischen Sequenzen enthält. Nach der post-transkriptionellen Verarbeitung werden die Introns herausgespleißt und ein polyA-Schwanz und eine 5′ – Kappe hinzugefügt, um reife mRNA-Transkripte zu erhalten, die in Proteine übersetzt werden können.,
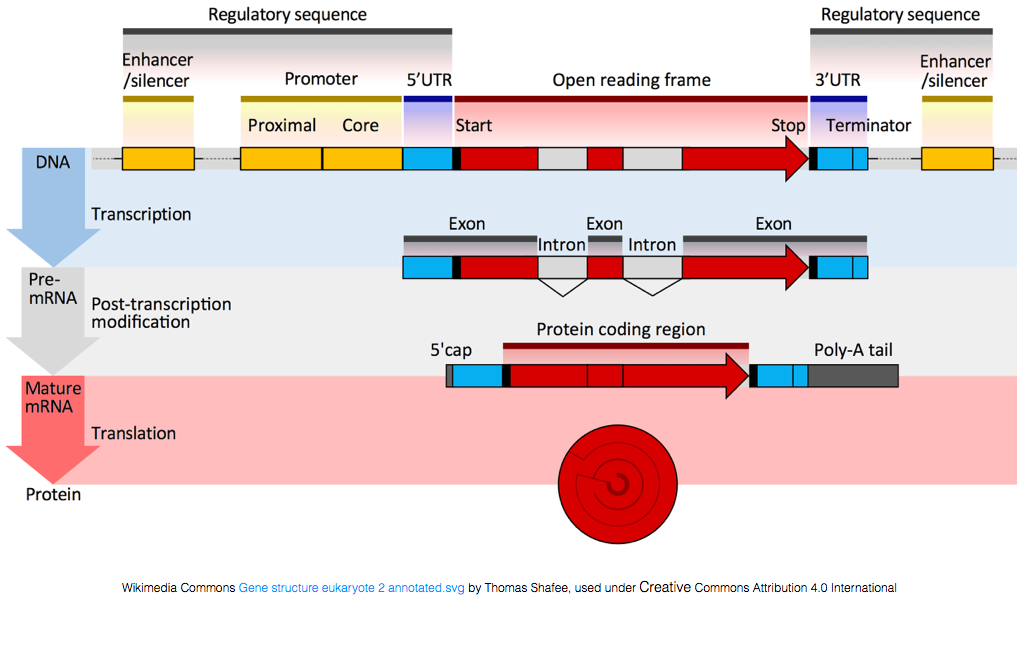
Während mRNA-Transkripte einen polyA-Schwanz haben, tun dies viele der nicht kodierenden RNA-Transkripte nicht, da die post-transkriptionelle Verarbeitung für diese Transkripte unterschiedlich ist.
Transkriptomik
Das Transkriptom ist definiert als eine Sammlung aller in einer Zelle vorhandenen Transkriptauslesungen., RNA-seq-Daten können verwendet werden, um das Transkriptom eines Organismus zu erforschen und/oder zu quantifizieren, das für die folgenden Arten von Experimenten verwendet werden kann:
- Differentielle Genexpression: quantitative Bewertung und Vergleich der Transkriptniveaus
- Transkriptomanordnung: Erstellung des Profils transkribierter Regionen des Genoms, qualitative Bewertung.,
- Kann verwendet werden, um bessere Genmodelle zu erstellen und diese mithilfe der Assembly
- Metatranscriptomics oder Community transcriptome analysis zu überprüfen
Illumina library preparation
Beim Starten eines RNA-seq-Experiments muss die RNA für jede Probe isoliert und in eine cDNA-Bibliothek für die Sequenzierung umgewandelt werden. Der allgemeine Workflow für die Bibliotheksvorbereitung ist in den folgenden Schritt-für-Schritt-Bildern detailliert beschrieben.
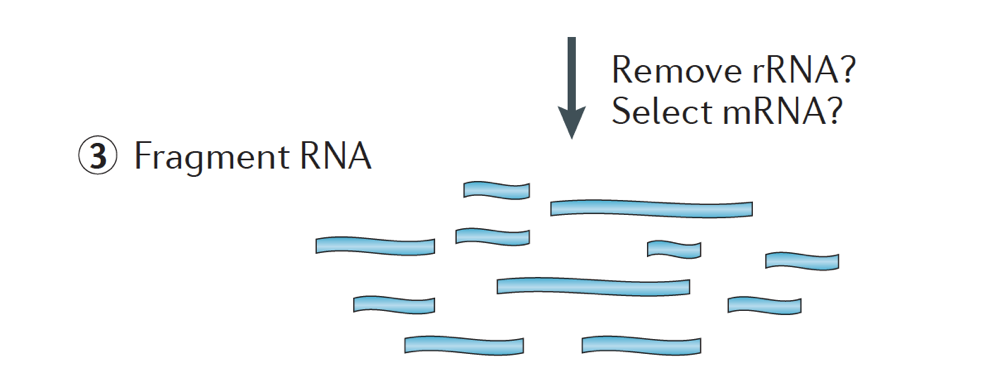
Kurz wird die RNA aus der Probe isoliert und kontaminierende DNA mit DNase entfernt.,
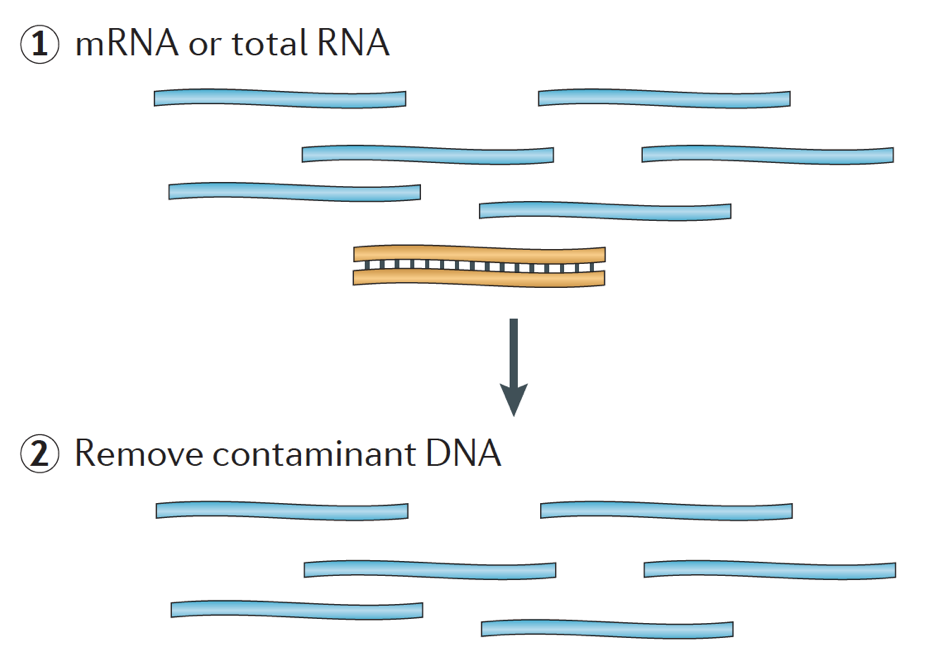
Die RNA-Probe wird dann entweder der Auswahl der mRNA (polyA-Auswahl) oder der Erschöpfung der rRNA unterzogen. Die resultierende RNA ist fragmentiert.
Im Allgemeinen repräsentiert ribosomale RNA die Mehrheit der in einer Zelle vorhandenen RNAs, während Messenger-RNAs einen kleinen Prozentsatz der gesamten RNA darstellen, ~2% beim Menschen. Wenn wir also die proteinkodierenden Gene untersuchen wollen, müssen wir uns für mRNA anreichern oder die rRNA abbauen., Für die differentielle Genexpressionsanalyse ist es am besten, sich für Poly(A)+ anzureichern, es sei denn, Sie möchten Informationen über lange nicht kodierende RNAs erhalten und führen dann eine ribosomale RNA-Erschöpfung durch.
Die Größe der Zielfragmente in der endgültigen Bibliothek ist ein Schlüsselparameter für die Bibliothekskonstruktion. Die DNA-Fragmentierung erfolgt typischerweise durch physikalische Methoden (d. H. akustisches Scheren und Sonieren) oder enzymatische Methoden (d. H. unspezifische Endonuklease) und Transposase-Fragmentierungsreaktionen.,
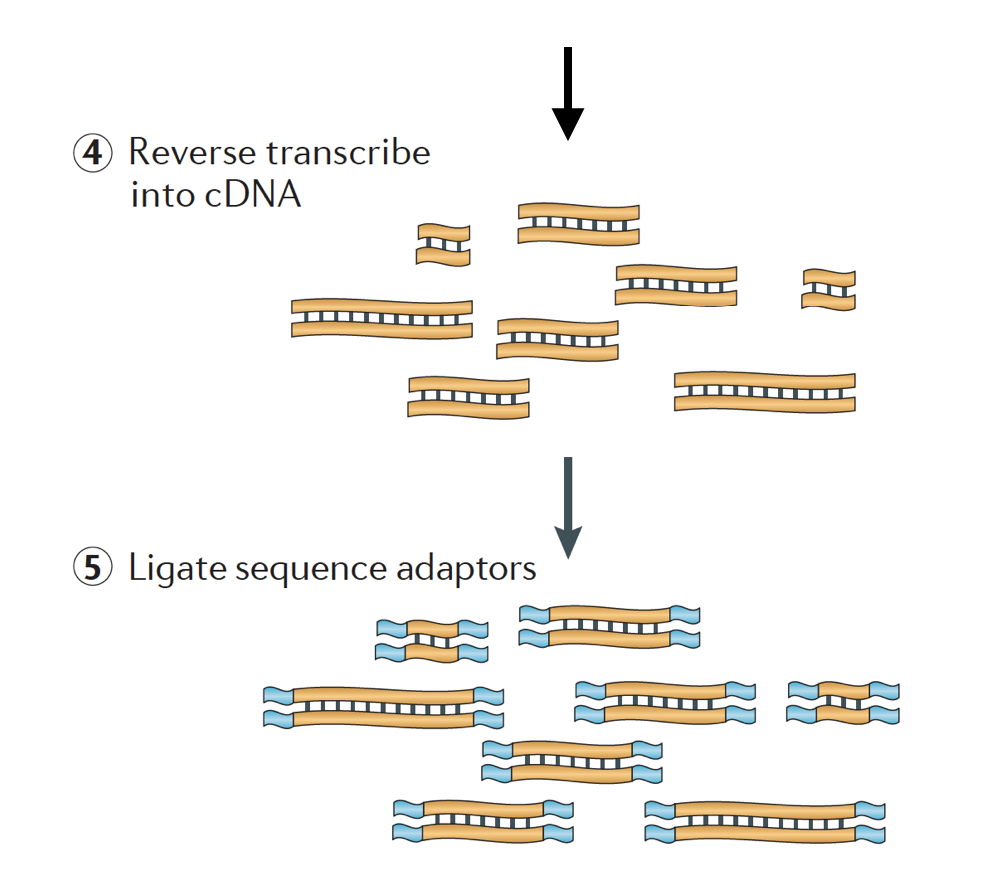
Die RNA wird dann rückwärts in doppelsträngige cDNA transkribiert und Sequenzadapter werden dann zu den Enden der Fragmente hinzugefügt.
Die cDNA-Bibliotheken können so erzeugt werden, dass Informationen darüber gespeichert werden, aus welchem DNA-Strang die RNA transkribiert wurde. Bibliotheken, die diese Informationen aufbewahren, werden Stranded Libraries genannt, die jetzt mit Illuminas TruSeq Stranded RNA-Seq Kits Standard sind., Gestrandete Bibliotheken sollten nicht teurer sein als nicht umbenannt, daher gibt es keinen Grund, diese zusätzlichen Informationen nicht zu erwerben.,
Es stehen 3 Arten von cDNA – Bibliotheken zur Verfügung:
- Forward (secondstrand) – Lesevorgänge ähneln der Gensequenz oder der Secondstrand-cDNA-Sequenz
- Reverse (firststrand) – Lesevorgänge ähneln dem Komplement der Gensequenz oder cDNA-Sequenz des ersten Ranges (TruSeq)
- Nicht eingeordnet
Schließlich werden die Fragmente bei Bedarf PCR-verstärkt und die Fragmente werden in der Größe ausgewählt (normalerweise ~300-500bp), um die Bibliothek zu beenden.
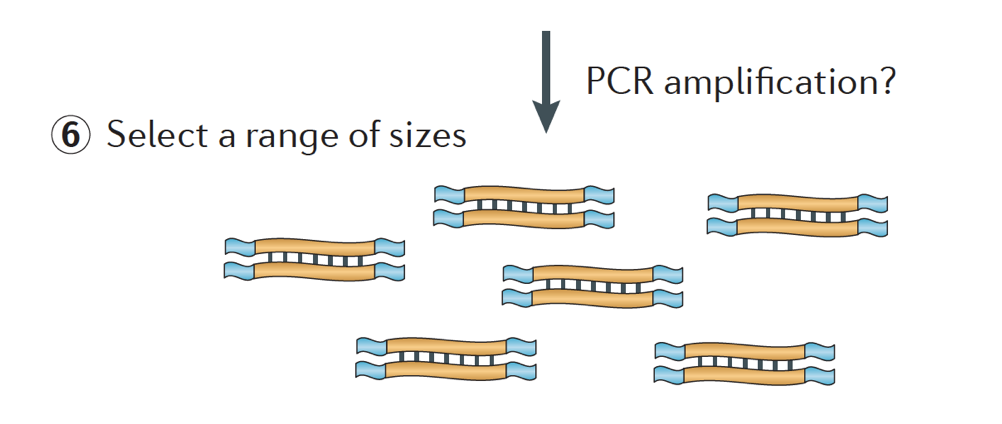
Bildnachweis: Martin J. A., Wang Z., Nat. Rev., Genet. (2011) 12:671-682
Illumina Sequencing
Single-end versus Paired-end
Nach Vorbereitung der Bibliotheken kann eine Sequenzierung durchgeführt werden, um die Nukleotidsequenzen der Enden der Fragmente zu erzeugen, die als Lesevorgänge bezeichnet werden. Sie haben die Wahl, ein einzelnes Ende der cDNA-Fragmente (Single-End-Lesevorgänge) oder beide Enden der Fragmente (Paarend-Lesevorgänge) zu sequenzieren.,
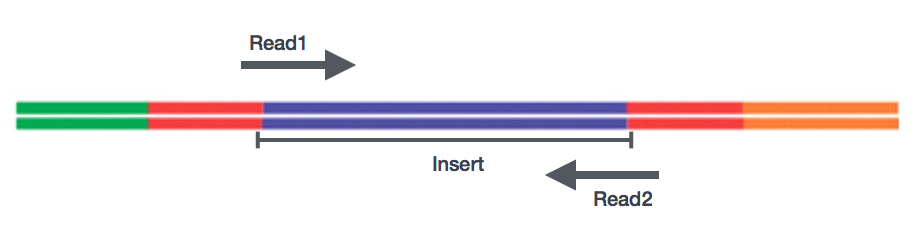
- SE – Single end dataset => Nur Read1
- PE – Paired-end dataset => Read1 + Read2
- kann 2 separate FastQ-Dateien oder nur eine mit verschachtelten Paaren sein
Im Allgemeinen ist eine Single-End-Sequenzierung ausreichend, es sei denn, es wird erwartet, dass die Lesevorgänge mit mehreren Stellen im Genom (z. B. Organismen mit vielen paralogen Genen), Baugruppen oder zur Spleiß-Isoform-Differenzierung übereinstimmen. Beachten Sie, dass High-End-Lesevorgänge im Allgemeinen 2x teurer sind.,
Verschiedene Sequenzierplattformen
Zur Sequenzierung der cDNA-Bibliotheken stehen verschiedene Illumina-Plattformen zur Auswahl.

Bildnachweis: Angepasst von Illumina
Unterschiede in der Plattform können die Länge der generierten Lesevorgänge, die Qualität der Lesevorgänge sowie die Gesamtzahl der Lesevorgänge, die pro Lauf sequenziert werden, und die Zeit ändern, die zum Sequenzieren der Bibliotheken erforderlich ist., Die verschiedenen Plattformen verwenden jeweils eine andere Flusszelle, bei der es sich um eine Glasoberfläche handelt, die mit einer Anordnung gepaarter Oligos beschichtet ist, die die Adapter ergänzen, die Ihren Vorlagenmolekülen hinzugefügt werden. In der Flusszelle finden die Sequenzierungsreaktionen statt.

Bildnachweis: Angepasst von Illumina
Sequencing-by-synthesis
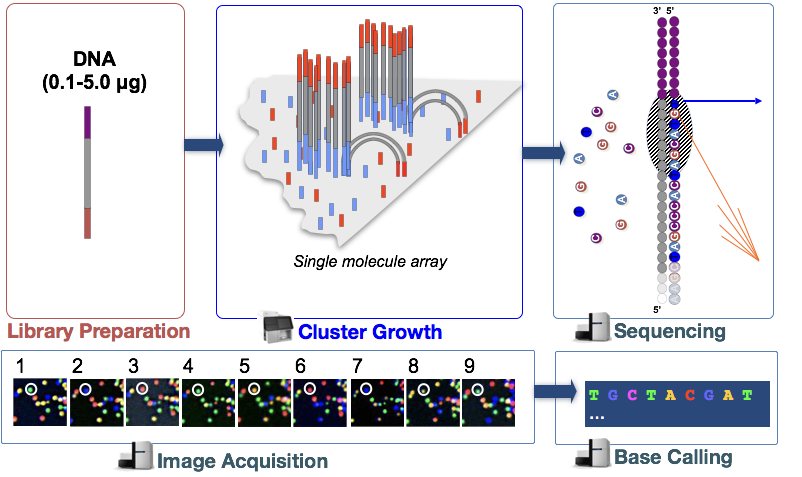
Illumina sequencing technology verwendet einen Sequencing-by-Synthesis-Ansatz, der im Folgenden näher beschrieben wird.
Im Schritt werden die DNA-Fragmente in der cDNA-Bibliothek denaturiert und auf die Glasflusszelle aufgebracht., Diese denaturierten Fragmente binden an die komplementären Oligos, die bereits kovalent an die Flusszellspuren gebunden sind, was zu einer Anheftung führt.
Clustergenerierung
Sobald die Fragmente angehängt sind, beginnt eine Phase namens Clustergenerierung. Während dieses Schritts werden einzelne Fragmente klonal verstärkt, um einen Cluster (Fragmente in unmittelbarer Nähe) identischer Fragmente zu erzeugen. Dies ist notwendig, damit die Fluoreszenz während des Nukleotideinbaus im nächsten Schritt von jedem Cluster anstelle eines einzelnen Fragments leicht erfasst werden kann.,
- Synthetisieren Sie das Komplement mit Polymerase
- dsDNA wird denaturiert, und die ursprüngliche DNA wird weggespült und der synthetisierte Strang kovalent an die Zelle gebunden.
- Einzelstranghybridisiert mit angrenzendem Adapter zu einer ‚Brücke‘
- dsDNA wird durch Polymerase erweitert. Jeder Strang kovalent an verschiedene Adapter gebunden.
- Wiederholen Sie viele Male, um alle eindeutigen Fragmente in der Zelle klonal zu verstärken und Cluster identischer Sequenz zu bilden.,
Sequenzierung durch Synthese (& Bilderfassung)
Nach der Clustergenerierung werden fluoreszenzmarkierte Nukleotide einzeln (zyklisch) eingebaut und Fluoreszenzbilder erfasst, um zu identifizieren, welches Nukleotid in jedem Zyklus in jeden Cluster integriert wird.
- Denaturcluster und der Block 3′ endet, um eine unerwünschte Grundierung zu verhindern.
- Hybridisieren Sie Sequenzierprimer zu Adaptersequenzen an den losen Enden.,
- Zyklus vier NTPs mit Fluoreszenzmarkern und Terminatorsequenz und Polymerasen.
- Sobald NTP integriert ist, wird der Cluster von einer Lichtquelle angeregt und ein charakteristisches fluorozentes Signal wird ausgesendet.
- Die Farbe wird aufgezeichnet, dann wird der Terminator auf Farbstoff gespalten und gewaschen. Prozesswiederholungen für bestimmte Anzahl von Zyklen.,
Base Calling
Illumina verfügt über eine proprietäre Software, die alle in der vorherigen Stufe aufgenommenen Bilder durchläuft und Textdateien mit Sequenzinformationen zu jedem Cluster basierend auf der Fluoreszenz generiert. Zusätzlich zum Aufrufen der Basen weist diese Software eine Wahrscheinlichkeitsbewertung zu, um anzuzeigen, wie sicher es war, etwas ein „A“, ein „T“, ein „G“ oder ein „C“aufzurufen.
Wenn es Unklarheiten gibt, z.B., bei einem bestimmten Zyklus hat das Bild für einen Cluster keine eindeutige Farbe, die einem bestimmten Nukleotid zugeordnet werden kann, die Basisaufrufsoftware hat eine geringe Wahrscheinlichkeit und weist ein „N“ anstelle von „A“, „T“, „G“ oder „C“zu.
Abschließend
- Anzahl der Cluster ~= Anzahl der Lesevorgänge
- Anzahl der Sequenzierungszyklen = Länge der Lesevorgänge
Die Anzahl der Zyklen (Länge der Lesevorgänge) hängt von der verwendeten Sequenzierungsplattform und Ihren Vorlieben ab.
ANMERKUNG., Wenn Sie die Sequenzierung durch Synthese genauer untersuchen möchten, empfehlen wir diese wirklich schöne Animation, die auf Illuminas YouTube-Kanal verfügbar ist.
Multiplexing
Abhängig von der Illumina-Plattform (MiSeq, HiSeq, NextSeq) variiert die Anzahl der Spuren pro Durchflusszelle und die Anzahl der Lesevorgänge, die pro Spur erhalten werden können, stark. Sie müssen entscheiden, wie viele Lesevorgänge Sie pro Sample (dh die Sequenztiefe) möchten, und dann basierend auf der Plattform berechnen, wie viele Gesamtspuren Sie für Ihren Sampleset benötigen., Wir werden mehr über Überlegungen sprechen, wenn Sie diese Entscheidung in der nächsten Lektion über experimentelle Überlegungen treffen
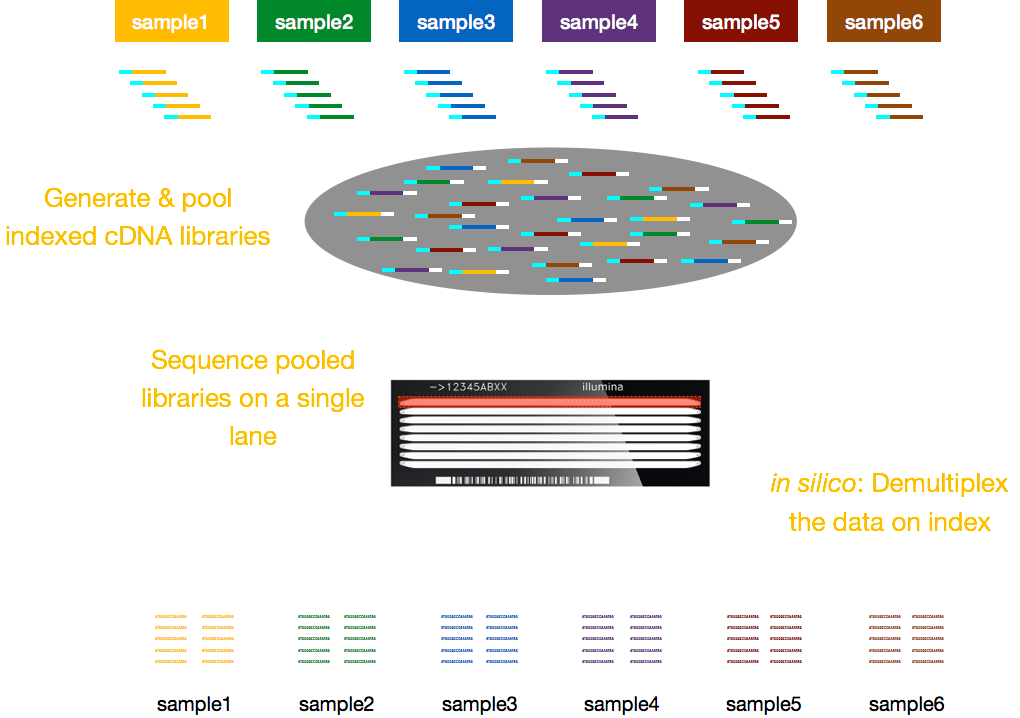
Typischerweise sind Gebühren für die Sequenzierung pro Spur der Flusszelle und Sie können mehrere Proben pro Spur ausführen. Illumina hat daher eine nette Multiplexmethode entwickelt, mit der Bibliotheken aus mehreren Proben gleichzeitig in derselben Spur einer Durchflusszelle gebündelt und sequenziert werden können. Diese Methode erfordert das Hinzufügen von Indizes (innerhalb des Illumina-Adapters) oder speziellen Barcodes (außerhalb des Illumina-Adapters), wie im folgenden Schema beschrieben.,
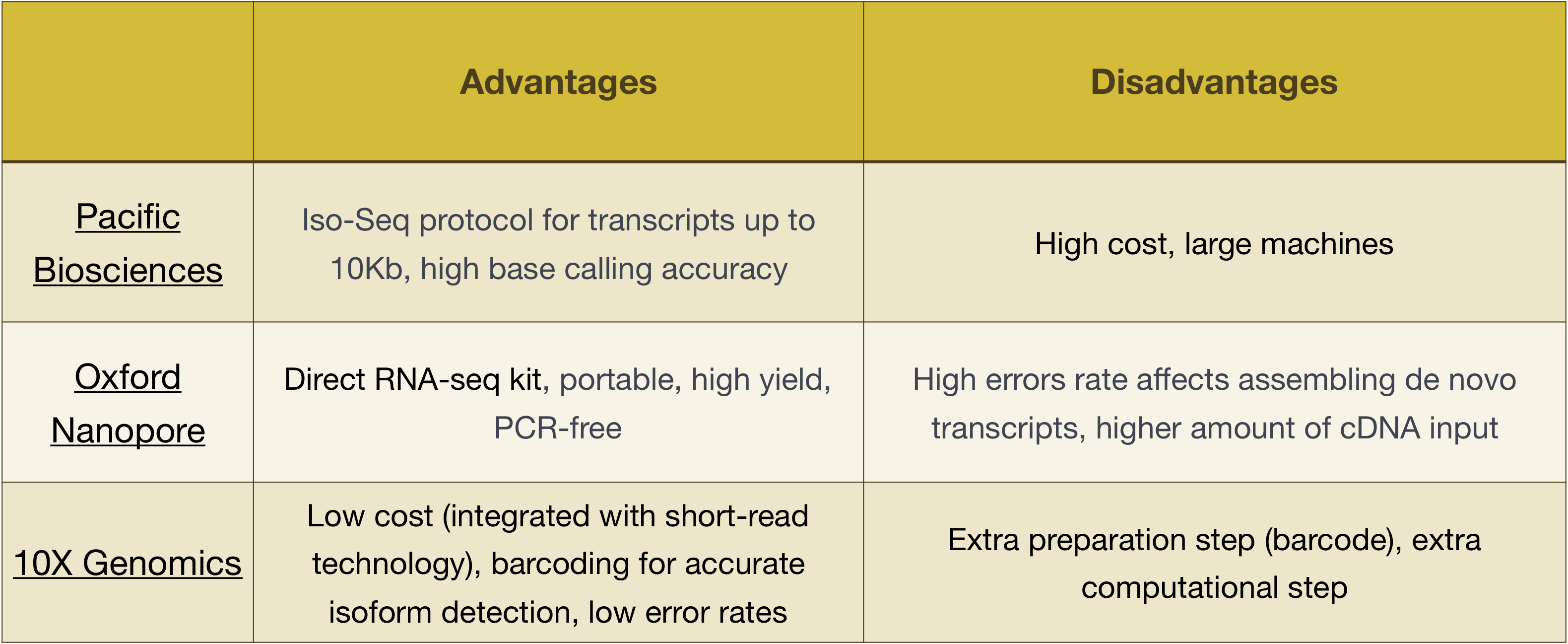
HINWEIS: Der in dieser Lektion vorgestellte Workflow ist spezifisch für Illumina sequencing, das derzeit die am häufigsten verwendete Sequenzierungsmethode ist., Aber es gibt auch andere long-read-Sequenzierung Methoden erwähnenswert, wie:
- Pacific Biosciences: http://www.pacb.com/
- Oxford Nanopore (MinION): https://nanoporetech.com/
- 10X Genomics: https://www.10xgenomics.com/
Vorteile und Nachteile dieser Technologien können erforscht werden, die in der folgenden Tabelle:
Schreibe einen Kommentar